J. Biosci. Public Health. 2025; 1(3)
Corresponding Author *
mdrakibulhasanbmb13.gb@gmail.com
![]() https://orcid.org/0009-0007-4355-0327
https://orcid.org/0009-0007-4355-0327
Affiliations:
Department of Biochemistry and Molecular Biology, Gono Bishwabidalay, Savar, Dhaka 1344, Bangladesh
Corresponding Author *
mdrakibulhasanbmb13.gb@gmail.com
![]() https://orcid.org/0009-0007-4355-0327
https://orcid.org/0009-0007-4355-0327
Affiliations:
Department of Biochemistry and Molecular Biology, Gono Bishwabidalay, Savar, Dhaka 1344, Bangladesh
Coauthor
mdrakibulhasanbmb13.gb@gmail.com
![]() https://orcid.org/0009-0007-4355-0327
https://orcid.org/0009-0007-4355-0327
Affiliations:
Department of Biochemistry and Molecular Biology, Gono Bishwabidalay, Savar, Dhaka 1344, Bangladesh
Coauthor
mdrakibulhasanbmb13.gb@gmail.com
![]() https://orcid.org/0009-0007-4355-0327
https://orcid.org/0009-0007-4355-0327
Affiliations:
Department of Biochemistry and Molecular Biology, Gono Bishwabidalay, Savar, Dhaka 1344, Bangladesh
Coauthor
mdrakibulhasanbmb13.gb@gmail.com
![]() https://orcid.org/0009-0001-1329-2255
https://orcid.org/0009-0001-1329-2255
Affiliations:
Department of Biochemistry and Molecular Biology, Gono Bishwabidalay, Savar, Dhaka 1344, Bangladesh
Coauthor
mdrakibulhasanbmb13.gb@gmail.com
![]() https://orcid.org/0009-0004-6410-3874
https://orcid.org/0009-0004-6410-3874
Affiliations:
Department of Biochemistry and Molecular Biology, Gono Bishwabidalay, Savar, Dhaka 1344, Bangladesh
Burkholderia pseudomallei (B. pseudomallei), the etiological agent of melioidosis, is present in the water and soil of equatorial and subtropical climates worldwide. Melioidosis occurs after environmental exposure to B. pseudomallei and is linked to comorbidities that impair the immune response, including diabetes, as well as socioeconomic hardship. This study aims to characterize and describe the functional annotation of the hypothetical protein of B. pseudomallei. The physicochemical properties determined the protein’s theoretical pI, estimated half-life in different media, and the negative hydropathicity of the selected protein. The subcellular identification determined that the protein was in the extracellular location with no transmembrane helices. The protein contained a functional domain associated with the aldose 1-epimerase superfamily that is related to carbohydrate metabolism. The protein-protein interaction network identified the protein’s correlation with 10 interacting proteins. The secondary structural documentation revealed that the protein contains mostly random coils, followed by extended strands and alpha helices. The tertiary structure modeling and assessment uncovered that the protein had a significant number of residues in the most favored regions compared to other regions. This functional protein can be targeted for further study on potential drug and vaccine candidates against the protein of B. pseudomallei.
Burkholderia pseudomallei (B. pseudomallei) is a type of intracellular bacterium found in the rhizosphere of equatorial soils. Melioidosis is caused by B. pseudomallei and is spread through contact with the skin, ingestion, or inhalation of contaminated water or soil [1]. B. pseudomallei infection can cause a number of clinical symptoms, such as pneumonia, infections of the skin, bones, joints, genitourinary system, and central nervous system, as well as parotid lesions in children [2, 3]. The pathogenicity of mammals is linked with the intracellular life cycle of B. pseudomallei, commencing with attachment and assimilation by host cells. B. pseudomallei has the capacity to infect many eukaryotic cells, comprising macrophages, monocytes, neutrophils, and nonphagocytic cells. Upon internalization, a type-3 secretion system (T3SSBsa) enables B. pseudomallei to evade the phagosome, allowing the bacterium to multiply within the cytoplasm [3]. The auto transporter protein BimA facilitates actin polymerization, allowing B. pseudomallei to propagate from cell to cell by actin-based motility. This mechanism, in conjunction with the function of a type-6 secretion system (T6SS-5), leads to the fusing of host membranes and the development of multinucleated giant cells. Capsule polysaccharides enhance virulence and facilitate evasion of host innate immunity [4, 5]. B. pseudomallei infections are difficult to treat due to their resistance to antimicrobials, which is caused by several efflux pumps and decreased outer membrane permeability. B. pseudomallei is now thought to be endemic in an extensive range of tropical and subtropical regions, including Africa, South America, the Middle East, Central America, and the Caribbean, in addition to its traditional hotspots in Southeast Asia and northern Australia. B. pseudomallei may survive in a variety of soil and climate circumstances across the world [1, 6].
B. pseudomallei inhalation or inoculation may induce significant disease. The ambient bacteria B. pseudomallei quickly become an aggressive intracellular pathogen that may spread throughout the body [7, 8]. The synthesis of numerous virulence factors at each intracellular infection stage speeds infection. B. pseudomallei uses the type III secretion system to escape host-cell cytotoxic activities in the phagosome following invasion or phagocytosis [9]. Many secreted virulence agents affect the host cell, while bacterial cells change their energy metabolism, allowing intracellular proliferation. Polymerizing host cell actin into "actin tails" drives B. pseudomallei to host cell membranes, where the type-VI secretion system forms multinucleated giant cells (MNGCs) for cell-to-cell dispersion [10]. B. pseudomallei inhabits saturated fields, stagnant water bodies, salty ecosystems, and human and animal samples [9]. Our knowledge of how B. pseudomallei adapts to varied environmental settings, including its change to a human host, is limited, resulting in enhanced virulence [9, 11]. Numerous components encoded in a vast genome enable the bacteria to endure hostile settings and kill intracellular pathogens [12-14]. Clinical and health departments in low-endemicity areas must recognize B. pseudomallei and improve diagnosis and reporting [13, 15].
This study employs a comprehensive in silico approach that includes physicochemical analysis, subcellular localization, functional annotation, protein–protein interaction, and active site prediction in order to fully characterize the hypothetical protein AAFM48_25015 of B. pseudomallei. In contrast to previous studies that focused mainly on known virulence factors, this work explores an unknown protein, which advances our understanding of the unidentified genetic elements of B. pseudomallei. This investigation provides new insights into the pathogen's virulence and adaptability by clarifying its possible functional roles and interaction patterns. It also may provide the basis for finding of new disease treatment or vaccine targets.
2.1. Flowchart and Tools Used in This Study
The whole methodology and bioinformatics tools used in this study are highlighted in Figure 1.
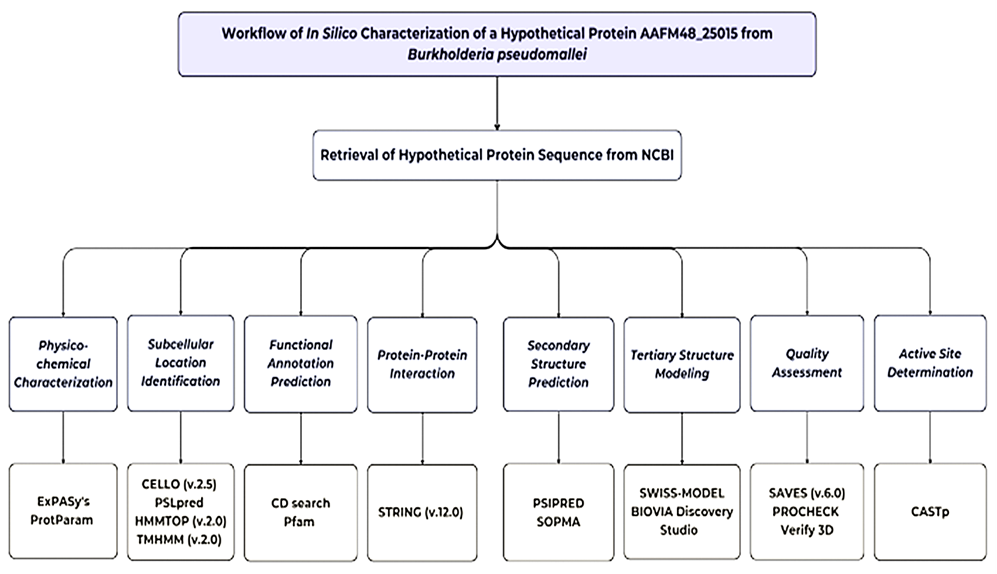
Figure 1. Complete workflow of in silico characterization of hypothetical protein AAFM48_25015 of B. pseudomallei
2.2. Sequence Retrieval
The amino acid sequence of the hypothetical protein of B. pseudomallei was retrieved in FASTA format from the National Center for Biotechnology Information (NCBI) with the accession ID of WZV79992.1, containing 207 amino acid residues [16]. This hypothetical protein AAFM48_25015 of B. pseudomallei was selected for this study.
2.3. Physicochemical Characterization
ExPASy's ProtParam tool was used to examine the target protein's physicochemical parameters, such as molecular weight, aliphatic index (AI), extinction coefficients, GRAVY (grand average of hydropathy), and isoelectric point (pI) [17].
2.4. Subcellular Location Identification
The subcellular location of the protein was documented by utilizing the CELLO (v.2.5) [18], PSLpred [19], HMMTOP (v.2.0) [20], and TMHMM (v.2.0) [21].
2.5. Functional Annotation Prediction
The NCBI platform’s CD search tool was utilized to predict the conserved domain in the protein AAFM48_25015 [22]. Protein motif determination was executed using the Pfamtool assigned to the evolutionary relationships of the protein AAFM48-25015 [23].
2.6. Protein-Protein Interaction
The STRING (v.12.0) program was used for determining the possible protein-protein interactions (PPI) [24].
2.7. Secondary Structure Prediction
The secondary structure of protein AAFM48_25015 has been predicted by the PSI-blast-based secondary structure prediction (PSIPRED) server-based tools, and the self-optimized prediction method with alignment (SOPMA) framework was used for element prediction [25, 26].
2.8. Tertiary Structure Modeling
The Protein Data Bank (PDB) currently does not contain an experimentally determined tertiary structure for AAFM48_25015 of B. pseudomallei (PDB). The 3D structure of the designated protein was determined using the SWISS-MODEL server based on homology modeling [27]. The server automatically performs BLASTp searches to identify templates for each protein sequence. The 3D model structure was visualized by BIOVIA Discovery Studio Visualizer [28].
2.9. Quality Assessment
The PROCHECK of the SAVES (v.6.1) program was performed to predict the Ramachandran plot and validate the predicted tertiary structure [29]. Also, verify the 3D of the SAVES (v.6.1) program used for verification of the 3D structure [30]. Additionally, the ERRAT server evaluated non-bonded atomic interactions to provide an overall quality factor [31].
2.10. Active Site Determination
We used the PrankWeb machine learning server to identify and rank possible active sites, which was used to carry out the prediction of potential ligand-binding pockets of the modeled protein [32].The amino acid sequence of the hypothetical protein of B. pseudomallei was retrieved in FASTA format from the National Center for Biotechnology Information (NCBI) with the accession ID of WZV79992.1, containing 207 amino acid residues [16]. This hypothetical protein AAFM48_25015 of B. pseudomallei was selected for this study.
3.1. Sequence Retrieval
The NCBI database was used to retrieve the hypothetical protein AAFM48_25015 from Burkholderia pseudomallei, as shown in Table 1. The protein has 207 amino acids (aa) and is classified as a hypothetical protein. It is identified by the locus WZV79992. The source organism has been identified as B. pseudomallei, and the accession number is WZV79992.1. The full amino acid sequence is given in FASTA format, and this will be the basis for further structural and functional characterization studies.
Table 1. Hypothetical protein AAFM48_25015 retrieval of B. pseudomallei from NCBI.
| Protein Individualities | Protein Information | |
| Locus | WZV79992 | |
| Amino acid | 207 aa | |
| Definition | hypothetical protein AAFM48_25015 | |
| Accession | WZV79992 | |
| Version | WZV79992.1 | |
| Source | Burkholderia pseudomallei | |
| Organism | Burkholderia pseudomallei | |
| FASTA sequence | MARLKAASLPPLITEPQHEHDLALYLLAPFSNRIGGGGFAWNGRRHDLRPNVEGEPCPLHGDAWQQHWTV MDQGRTHVTLQLLSRSIPPFAYDAEITWTLDGPSLSGALTLIHRGEDPMPYGGGFHPWLVRDSDTQLHAR ADGWWSEDSLHLPKRWHALVDNDSSGFGAPRSLPSNFSIRSTRAGTVERTSGGQGAALLFRSRLRRP | |
3.2. Physicochemical Characterization
The hypothetical protein AAFM48_25015 has a molecular weight of 23005.81 Da and a sequence of 207 amino acids. When all Cys residue pairs form cystines, the extinction coefficient is 14105; when all Cys residues are reduced, the extinction coefficient is 13980. The protein has 21 negatively charged residues (Asp + Glu) and 21 positively charged residues (Arg + Lys), making it partially basic (pI 7.21). Table 2 shows that the aliphatic index, instability index, and GRAVY are 75.02, 58.25, and -0.514, respectively.
Table 2. Physicochemical characterization of hypothetical protein AAFM48_25015.
| Physicochemical Parameters | Values |
| Number of amino acids | 207 |
| Molecular weight | 23005.81 |
| Theoretical isoelectric point (pI) | 7.21 |
| Formula | C1024H1564N308O293S4 |
| Total number of atoms | 3193 |
| The estimated half-life | a) about 30 hours (mammalian reticulocytes, in vitro). b) >20 hours (yeast, in vivo). c) >10 hours (Escherichia coli, in vivo) |
| Aliphatic index | 75.02 |
| Instability index | 58.25 |
| Extinction coefficients (all pairs of Cys subunits form cystines) | 48470 |
| Extinction coefficients (all Cys subunits are reduced) | 48470 |
| Total number of negatively charged components (Asp + Glu) | 21 |
| Total number of positively charged residues (Arg + Lys) | 21 |
| Grand average of hydropathicity (GRAVY) | -0.514 |
3.3. Subcellular Location Identification
The CELLO (v.2.5) and PSLpred tools were utilized for subcellular location assessment of the protein AAFM48_25015. The tools predicted the subcellular location of the protein as an extracellular protein. The HMMTOP (v.2.0) and TMHMM (v.2.0) algorithms predicted that the protein AAFM48_25015 did not contain any transmembrane helices, highlighting the protein's extracellular location within B. pseudomallei, as is apparent in Table 3.
Table 3. Subcellular location of hypothetical protein AAFM48_25015.
| Analysis Tools | Result |
| CELLO (v.2.5) | Extracellular (1.210) |
| PSLpred | Extracellular |
| HMMTOP (v.2.0) | No transmembrane helices |
| TMHMM (v.2.0) | No transmembrane helices |
3.4. Functional Annotation Prediction
A domain of a protein is a conserved part of a given protein sequence that has a function, and it exists independently of the rest of the protein chain. In Figure 2A, CD search program assumed a conserved domain (accession ID: cl14648) belonging to the Aldose 1-epimerase superfamily, with sequence alignment highlighting homology to related protein families. As shown in Figure 2B, Pfam motif and protein family analysis further predicted the presence of the Aldose 1-/Glucose-6-phosphate 1-epimerase domain (accession ID: IPR008183), supporting its functional classification within the Aldose epimerase family, which is commonly associated with carbohydrate metabolism, particularly the interconversion of α- and β-anomers of aldose sugars.
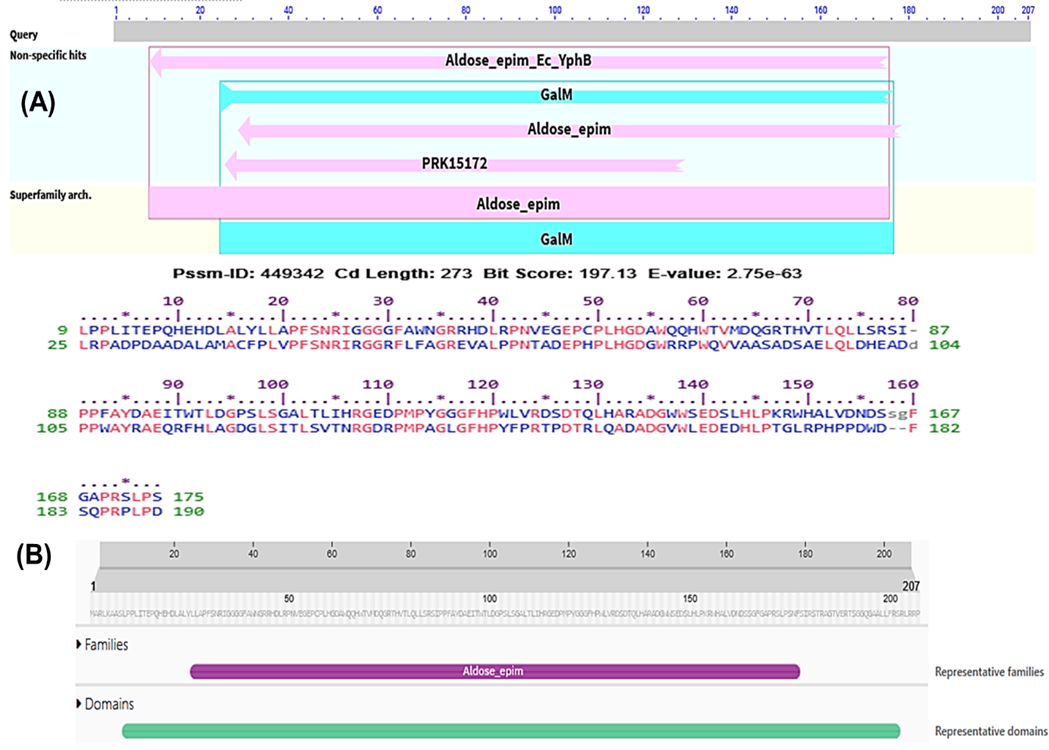
Figure 2. Functional annotation for the protein AAFM48_25015. The graphical summary depicts the conserved domains detected in the inquiry sequence. The aligned sequences show the conserved domains found on the searched sequence and contrast them to the homologous protein domain family, Aldose 1-epimerase superfamily (CDD accession no. cl14648) (A), and Pfam motif search tools predicted the Aldose 1-/Glucose-6-phosphate 1-epimerase domain (IPR008183) of the protein AAFM48_25015 (B).
3.5. Protein-Protein Interaction
The STRING v.12.0 tool was used to ascertain the protein-protein interaction (PPI) that resulted when Aldose 1-epimerase (BPSS2067) interacted with 10 proteins as displayed in Figure 3.
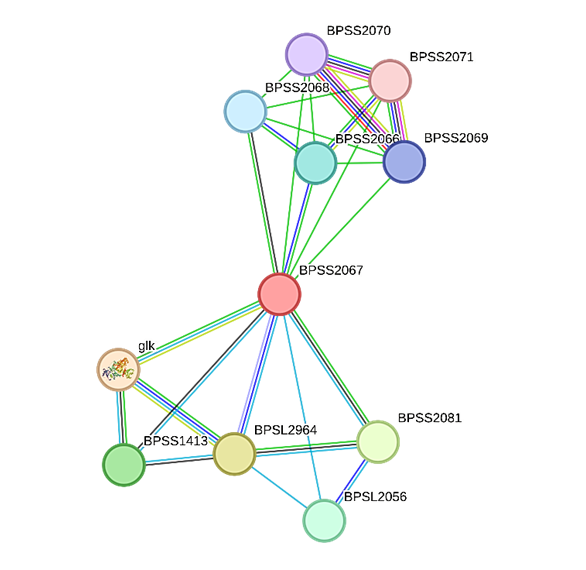
Figure 3. The protein's STRING system regulates protein-protein interactions. The colored nodes represent the desired proteins and the initial shell of interaction sources. Regarding cluster material, vacant clusters indicate proteins with unknown 3D structures, and full nodes reflect known or predicted 3D structures.
The list of proteins identified using the STRING database that are predicted to interact with Aldose 1-epimerase (BPSS2067) in Burkholderia pseudomallei is shown in Table 4. Strong relationships with enzymes involved in the metabolism of carbohydrates, such as glucokinase (glk) and a putative epimerase, are highlighted by the interaction network; both have high confidence scores (>0.90). The presence of alpha-galactosidase (RafA), putative hydrolases, oxidases, and short-chain dehydrogenases as additional predicted interactors may indicate that Aldose 1-epimerase plays a functional role in redox and sugar utilization. Numerous hypothetical proteins and transport-related proteins (such as ribose transport permease and ABC transporter components) were also found, suggesting a more extensive role in substrate transport and metabolic regulation. All of these interactions point to the possibility that Aldose 1-epimerase is a part of a complex metabolic network that is essential to B. pseudomallei's adaptability and pathogenic potential.
Table 4. List of proteins that interact with Aldose 1-epimerase (BPSS2067) identified through STRING, with their short descriptions.
| Interacting Proteins | Short Descriptions | Score |
| glk | Glucokinase | 0.917 |
| BPSL2964 | Putative epimerase | 0.915 |
| BPSS2081 | Alpha-galactosidase RafA | 0.907 |
| BPSS1413 | Putative hydrolase protein | 0.901 |
| BPSL2056 | Putative oxidase | 0.900 |
| BPSS2066 | Similar to Yersinia pestis hypothetical protein ypo1289 SWALL: Q8ZGK2 | 0.820 |
| BPSS2068 | Short chain dehydrogenase | 0.774 |
| BPSS2069 | Similar to Rhizobium loti ABC transporter, ATP-binding protein mll7373 SWALL: Q986G1 | 0.752 |
| BPSS2070 | Similar to Escherichia coli ribose transport system permease protein RbsC SWALL: RBSC_ECOLI | 0.752 |
| BPSS2071 | Putative exported protein | 0.700 |
3.6. Amino Acid Composition and Secondary Structure Prediction
The amino acid composition of the hypothetical protein AAFM48_25015 was obtained from the ExPASy ProtParam Tool is shown in Table 5. The protein consists of 207 amino acids, with leucine (12.1%) being the most abundant residue, followed by glycine (10.1%), and arginine, serine, proline, and alanine (each 8–9%). In contrast, residues such as cysteine (0.5%), lysine (1.0%), methionine (1.4%), and tyrosine (1.4%) were present in low proportions. The balanced distribution of hydrophobic and polar residues suggests a stable structural framework, while the enrichment of leucine and glycine may contribute to the protein’s folding dynamics and functional flexibility.
Table 5. The amino acid profile derived from the hypothetical protein AAFM48_25015.
| SL No. | Amino Acids | No. of Amino Acids | Percentage |
| 1 | Ala (A) | 17 | 8.2 % |
| 2 | Arg (R) | 19 | 9.2 % |
| 3 | Asn (N) | 5 | 2.4 % |
| 4 | Asp (D) | 13 | 6.3 % |
| 5 | Cys (C) | 1 | 0.5 % |
| 6 | Gln (Q) | 7 | 3.4 % |
| 7 | Glu (E) | 8 | 3.9 % |
| 8 | Gly (G) | 21 | 10.1 % |
| 9 | His (H) | 11 | 5.3% |
| 10 | Ile (I) | 6 | 2.9 % |
| 11 | Leu (L) | 25 | 12.1 % |
| 12 | Lys (K) | 2 | 1.0% |
| 13 | Met (M) | 3 | 1.4% |
| 14 | Phe (F) | 7 | 3.4 % |
| 15 | Pro (P) | 17 | 8.2 % |
| 16 | Ser (S) | 17 | 8.2 % |
| 17 | Thr (T) | 11 | 5.3 % |
| 18 | Trp (W) | 8 | 3.9 % |
| 19 | Tyr (Y) | 3 | 1.4 % |
| 20 | Val (V) | 6 | 2.9 % |
The PSIPRED program shows higher confidence in the prediction of the helix, strand, and coil shown in Figure 4A. SOPMA results for the secondary structure modeling displayed the distribution of helix (blue), sheet (purple), and coil (orange) structures across the protein sequence in Figure 4B.
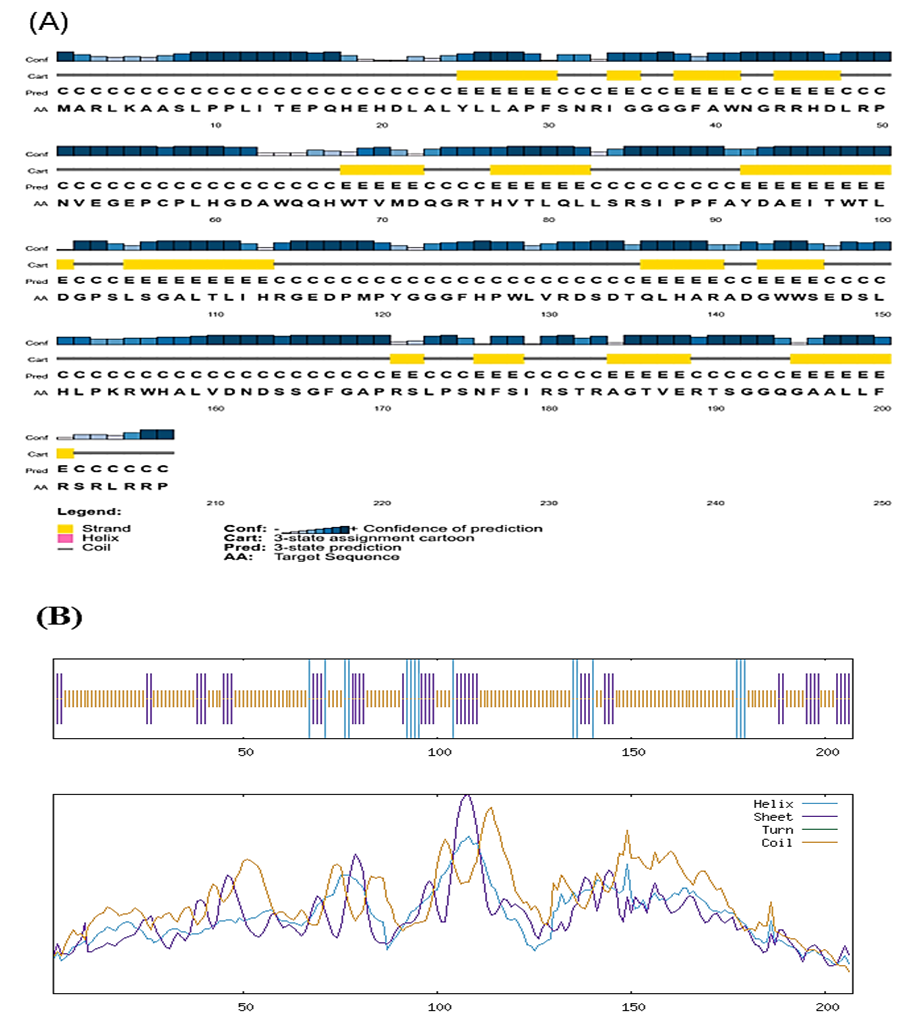
Figure 4. Secondary structure modeling from the hypothetical protein AAFM48_25015 using PSIPRED (A) and SOPMA service (B).
The secondary structural composition of the hypothetical protein AAFM48_25015 is summarized in Table 6. The analysis revealed that the protein is predominantly composed of random coils (72.95%), followed by extended β-strands (20.77%) and a small fraction of α-helices (6.28%). No 3<sub>10</sub> helices, π-helices, β-bridges, β-turns, or bend regions were detected. The predominance of random coils suggests that the protein may possess a flexible and dynamic structure, while the presence of extended strands and limited α-helical content indicates the potential formation of β-sheet-rich regions that could contribute to structural stability and functional specificity.
Table 6. Secondary structural elements of hypothetical protein AAFM48_25015.
| Secondary structural components | Values (%) |
| Alpha helix | 6.28 |
| 310 helixes (Gg) | 0.00 |
| Pi helix (Ii) | 0.00 |
| Beta bridge (Bb) | 0.00 |
| Extended strand (Ee) | 20.77 |
| Beta turn (Tt) | 0.00 |
| Bend region (Ss) | 0.00 |
| Random coil (Cc) | 72.95 |
| Ambiguous states | 0.00 |
| Other states | 0.00 |
3.7. Tertiary Structure Modeling
The tertiary structure of the corresponding protein was generated from the SWISS-MODEL service using the template A0A0F6L4K3.1.A, which has 91.49% sequence similarity with the target protein. Figure 5 shows how the SWISS-MODEL-generated modeling structure is represented in BIOVIA Discovery Studio.
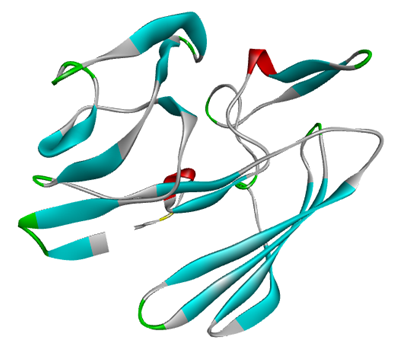
Figure 5. The target protein's 3D structure was predicted utilizing the SWISS-MODEL service and then displayed in BIOVIA Discovery Studio software.
3.8. Quality Assessment
The PROCHECK tool of the SAVESv6.1 server was used to evaluate the structural quality of the simulated protein. Figure 6A shows the configuration of the ψ and φ angles. Residues in the most desired locations wrapped 88.1% of the protein, confirming the predicted 3D structure (AAFM48_25015). Furthermore, residues in further sanctioned areas, bountifully authorized locations, and banned regions were all 0 (0.00%), whereas the number of nonglycine and nonproline residues and end residues (excluding Gly and Pro) were 151 (100.0%) and 1, respectively. Subsequently, the ERRAT overall quality factor of 91.837% indicates that the model is of high quality and structurally reliable for further analysis, as shown in Figure 6B. Finally, the Verify 3D program of the SAVES server showed this modeled structure quality, as 95.21% of the residues have an average 3D-1D score >= 0.1, as shown in Figure 6C.
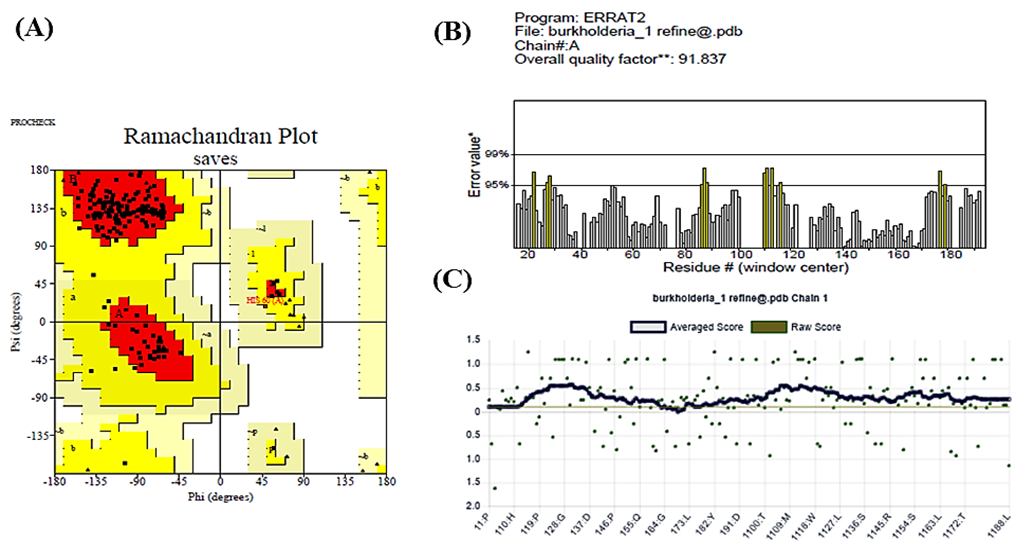
Figure 6. The PROCHECK software confirmed the statistics of the Ramachandran plot for the simulated three-dimensional structure (A), the ERRAT showed quality score(B), and the Verify 3D program showed the quality of this modeled structure (C).
Table 7 summarizes the Ramachandran plot statistics for the simulated protein, detailing the conformational quality of its backbone dihedral angles. Among the 151 non-glycine and non-proline residues, 88.1% (133 residues) found in the most favored regions, indicating a high-quality structure with stable conformations. In addition, 11.9% (18 residues) are in allowed regions, while no residues are found in generously allowed or disallowed regions, suggesting excellent stereochemical integrity. The protein includes one end-residue (excluding glycine and proline), 21 glycine residues, and 15 proline residues, with glycine residues represented as triangles in the plot. These results confirm the structural reliability of the simulated protein model.
Table 7. Ramachandran plot statistics of the simulated protein.
| Ramachandran plot statistics | Values (%) |
| Residues in most favored regions | 133 (88.1%) |
| Residues in additional allowed regions | 18 (11.9%) |
| Residues in generously allowed regions | 0 (0.00%) |
| Residues in disallowed regions | 0 (0.00%) |
| Number of non-glycine and non-proline residues | 151 (100.0%) |
| Number of end-residues (excl. Gly and Pro) | 1 |
| Number of glycine residues (shown as triangles) | 21 |
| Number of proline residues | 15 |
3.9. Active Site Determination
Figure 7 illustrates the modeled protein structure. The Prank Web server identified four unique ligand-binding pockets in the modeled protein. The pockets were delineated according to geometric and physicochemical characteristics, suggesting probable locations for ligand interaction and enzymatic activity. The protein is color-coded to distinguish different domains, with arrows indicating key structural features, providing a visual representation of the active sites critical for its function.
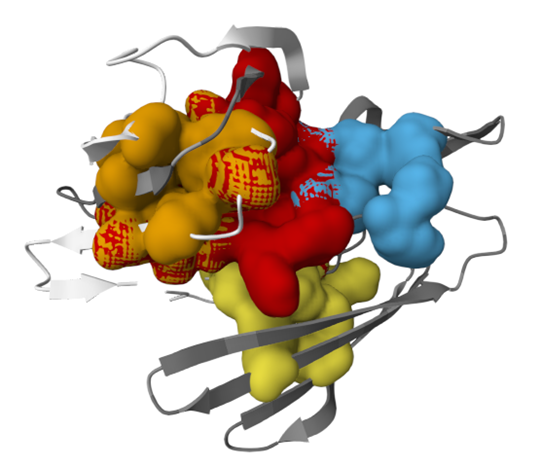
Figure 7. PrankWeb server prediction of four potential ligand-binding sites in the modeled protein, indicating possible regions for molecular interaction and functional activity.
In this study, in silico analysis of the theoretical protein AAFM48_25015 from Burkholderia pseudomallei provides valuable insights into its physicochemical properties and biological activities. The FASTA sequence study shows the molecular weight of this protein is 23,005.81 Da, with extinction coefficients of 14,105 (all cysteine residues converted to cystines) and 13,980 (all cysteine residues reduced), characterized by moderate capacity for light absorption [33]. The calculated isoelectric point (pI) of 7.21 indicates it is a weakly basic protein, which determines its solubility and electrophoretic mobility and is crucial while undergoing purification processes [17]. The aliphatic index (75.02) and instability index (58.25) suggest a moderately stable protein structure. The negative GRAVY score (-0.514) also confirms its hydrophilic nature, favoring an extracellular localization [34, 35]. The predicted half-life of over 10 hours in E. coli showsits resilience in varying media as well as validates its environmental flexibility [36].
The extracellular nature of the protein was confirmed by subcellular localization anticipated from CELLO (v.2.5) and PSLpred. Moreover, HMMTOP and TMHMM did not detect any transmembrane helices. Since adhesion and nutrient uptake are essential for survival of B. pseudomallei, this extracellular positioning implies a possibility that it plays an active role in external interactions [18, 19]. Pfam's prediction of the Aldose 1-/Glucose-6-phosphate 1-epimerase domain (IPR008183) supported the functional annotation using the CD Search program, which found a conserved domain (accession ID: cl14648) within the Aldose 1-epimerase superfamily. These enzymes are crucial for the metabolism of carbohydrates because they allow the α- and β-anomers of hexose sugars, such as glucose and galactose, to be interconverted, potentially increasing the metabolic flexibility of the bacterium [22, 23].
In addition, the protein-protein interaction (PPI) network analysis exhibits that Aldose 1-epimerase (BPSS2067) has ten interacting partners, including glucokinase and alpha-galactosidase, suggesting that it is integrated into a network of carbohydrate metabolism [24]. A flexible structure appropriate for dynamic interactions was suggested by the secondary structure modeling conducted by PSIPRED and SOPMA, which showed a large number of random coils with minimal α-helices and extended β-strands similar to the previous reports [25, 26]. The tertiary structure was modeled using a 91.49% identical sequence template (A0A0F6L4K3.1.A). The Ramachandran plot statistics showed 88.1% of residues in the most favored regions with no disallowed conformations [29-31, 37]. Beyond this, the discovery of four ligand-binding pockets on the PrankWeb server provides more credibility to its potential as an enzymatic or binding site, providing chances for the development of specific treatments [32]. Moreover, the extracellular localization and potential ligand-binding sites also raise the possibility of its involvement in host-pathogen signaling pathways; for instance, a study on the roles of Src family kinases in epidermal homeostasis and wound healing could be possible for therapeutic benefit [38].
The overall findings of this study may provide a robust foundation as well as experimental validation through enzymatic assays or molecular dynamics simulations to confirm its biological role and therapeutic potential roles in B. pseudomallei.
This study offers an in silico characterization of a hypothetical protein from Burkholderia pseudomallei, the causative agent of melioidosis, a globally underreported disease linked to environmental and climate changes. The protein's physicochemical properties, including its theoretical isoelectric point, estimated half-life, and negative hydropathicity, were determined. Subcellular localization analysis predicted the protein to be extracellular, with no transmembrane helices, and its functional annotation identified a domain linked with the aldose 1-epimerase superfamily, suggesting a role in carbohydrate metabolism. Protein-protein interaction analysis underlined associations with 10 interacting proteins, implying its involvement in metabolic pathways. Structural analyses revealed the protein predominantly comprises random coils, extended strands, and alpha helices, with favorable tertiary structure attributes. Although it has potential as a therapeutic or vaccine target, the study's reliance on computational predictions underscores the need for experimental validation. Further investigations are essential to confirm the protein's biological role and therapeutic significance.
The authors would like to thank the Department of Biochemistry and Molecular Biology, Faculty of Health Sciences, Gono Bishwabidyalay (University), Dhaka-1344, Bangladesh, for the technical support of this research. This study address about 11% AI assisted text or reference formatting.
This research was conducted with self-funding. Therefore, any kind of financial support was not received for this study.
The authors declare no conflicts of interest.
This study did not involve any experiments on human participants or animals; therefore, formal written informed consent was not required. All figures in this study were created; therefore, no permission for reuse is required for any figure presented herein. The authors confirm that all listed co-authors have significantly contributed to this work, and no other individuals were involved in carrying out this project. The responsibility for the content of this manuscript lies entirely with the authors.
 access
access