The epidermis is primarily composed of keratinocytes, maintain the integrity of skin by carefully controlling the ratio of differentiation to proliferation. Non-receptor tyrosine kinases known as Src family kinases (SFKs), which include Src, Fyn, and Yes, are essential for these processes because they affect tumorigenesis, wound healing, and epidermal maintenance. Through their interactions with receptor tyrosine kinases like EGFR, SFKs modulate keratinocyte migration, differentiation, and proliferation by activating downstream Ras-MAPK and PI3K-Akt pathways. Using integrin-mediated focal adhesion kinase (FAK) activation and cytoskeletal remodeling, SFKs facilitate keratinocyte migration during wound healing. By altering cell adhesion, modifying the epithelial–mesenchymal transition, and abnormally activating proliferative drivers like Yes-associated protein (YAP), dysregulation or overexpression of SFKs disrupts epidermal homeostasis and contributes to carcinogenesis. The development of cutaneous squamous cell carcinoma may be prevented or lessened by targeted inhibition of SFKs, which can be achieved with drugs such as dasatinib, bosutinib, AZD-0530, and bioactive phytochemicals. This review highlights some reported outcomes on the therapeutic approaches targeted at regaining epidermal function, enhancing wound healing, and slowing the spread of cancer while outlining present knowledge of SFK-mediated regulation of keratinocyte biology. Obtaining insight into SFK signaling pathways sets the basis for developing treatments for skin cancers and pathological wound healing.
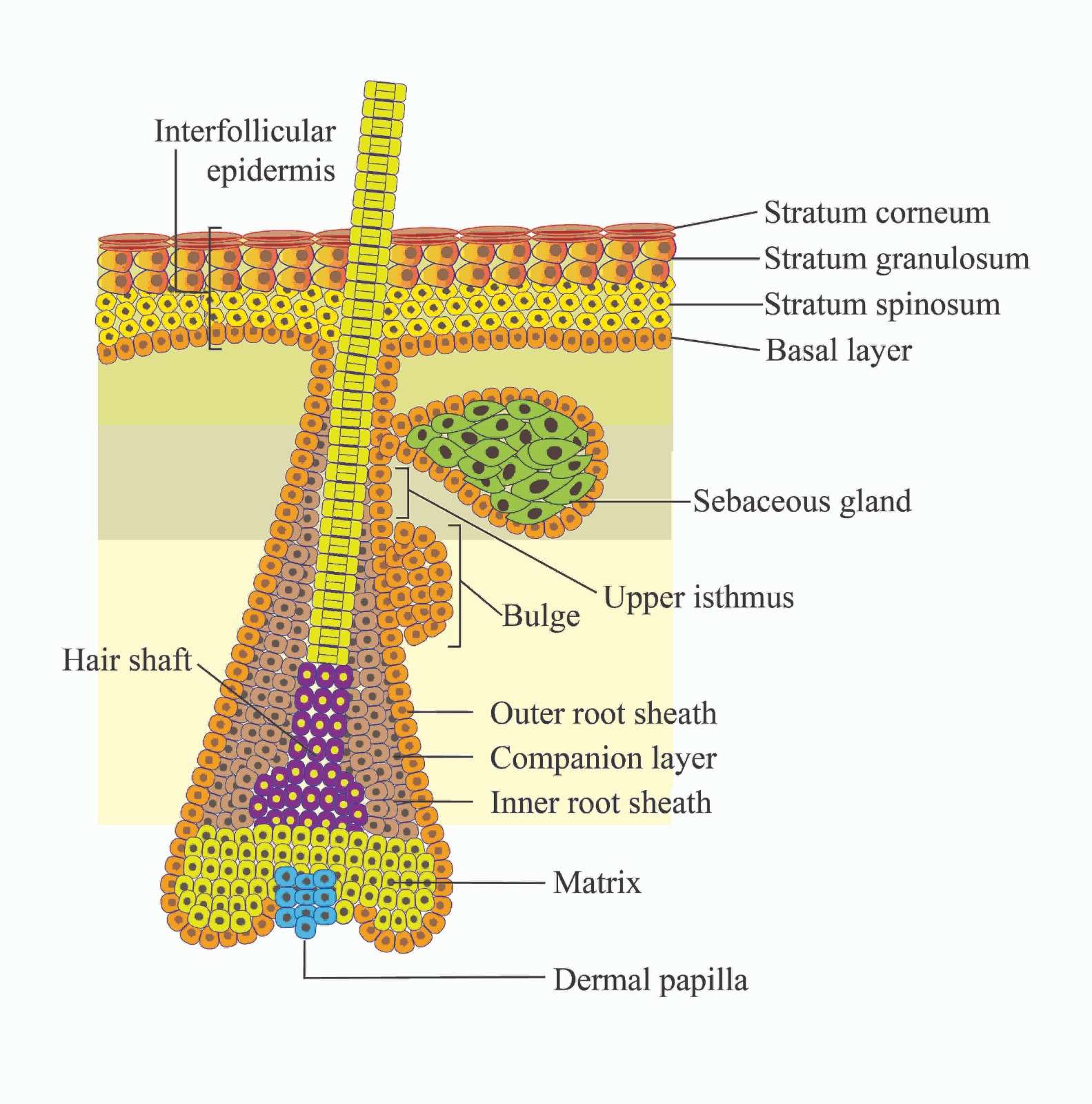
The epidermis, the outermost covering of the skin, protects our bodies from external elements and prevents dehydration. It is composed of stratified epithelial tissue that serves as a barrier against adverse environmental conditions and mainly consist of keratinocytes [1]. This squamous epithelial tissue is separated into two parts: the interfollicular epidermis (IFE) and distributed pilosebaceous units, which are termed appendages, such as the follicle that produces hair and the sebaceous gland that lubricate the skin [2]. These appendages invade the dermis from the IFE, and stem cells, or progenitor cells, make up nearly all cells in the inner layer of the IFE base. They regenerate themselves continuously. To produce the outer layers, which include the stratum spinosum (SS), stratum granulosum (SG), and stratum corneum (SC) layers, they move upward and undergo a highly structured differentiation process. The outermost layer, known as the SC, provides the skin’s primary barrier function and is made up of corneocytes encased in a lipid matrix that are dead, and keratin filled [3]. SCs are continuously removed from the tissue under healthy conditions and then replaced by other differentiating layer cells [4].
The three stages of a hair follicle are typically three years long: the anagen, or growth phase; the several-week-long catagen, or involuting phase; and the several-month-long telogen or resting phase. The bulge, which is the lowest portion of the enduring part of the hair follicle, is home to stem cells that are responsible for follicle renewal in hair during anagen. The transit-amplifying cells of the hair follicle are produced by the bulge stem cells during anagen. These multiply quickly and transiently before starting seven differentiation programs that eventually result in the adult hair follicle (Figure 1). Hair growth ceases after the matrix cells have used up all their proliferative potential, and the follicle enters catagen, resulting in the bulging portion of the follicle remaining intact while the lower two-thirds disintegrate. The lowest portion of the hair follicle contains the hair bulb. This structure is composed of differentiated offspring of the stem cells of the bulge and sit atop the dermal papilla. The dermal papilla contains specialized dermal fibroblasts, nerve fibers, and a capillary loop (Figure 1). It is essential for hair follicle growth and adult hair cycle regulation [5].
Complex signaling networks regulate keratinocyte growth and differentiation equilibrium. Among these, Src family kinases (SFKs), nonreceptor tyrosine kinases, play a crucial role in epidermal homeostasis. The dysregulation of SFKs impairs the healing process [6, 7], and overexpression promotes carcinogenesis [8]. This study summarizes the functions of Src family kinases, which control cancer formation, wound recuperation, and epidermal maintenance complex signaling networks regulate keratinocyte growth and differentiation equilibrium. Among these, Src family kinases (SFKs), nonreceptor tyrosine kinases, play a crucial role in epidermal homeostasis. Given their central role in both physiological and pathological contexts, SFKs represent promising therapeutic targets. Pharmacological inhibitors-including dasatinib, bosutinib, and AZD-0530—as well as bioactive phytochemicals have shown potential in modulating SFK activity to restore epidermal function, enhance wound healing [6,7], and limit tumor progression [9]. Understanding the molecular mechanisms by which SFKs regulate keratinocyte biology can inform the development of targeted therapies for skin disorders.
This mini-review synthesizes current knowledge on SFK-mediated regulation of epidermal maintenance, wound healing, and tumorigenesis, highlighting therapeutic strategies aimed at optimizing skin repair and preventing malignant transformation.
Figure 1. Structure of the epidermis of the skin.
2. OVERVIEW of Src FAMILY KINASES (SFKs)
2.1. Structure of SFKs and Their Activation
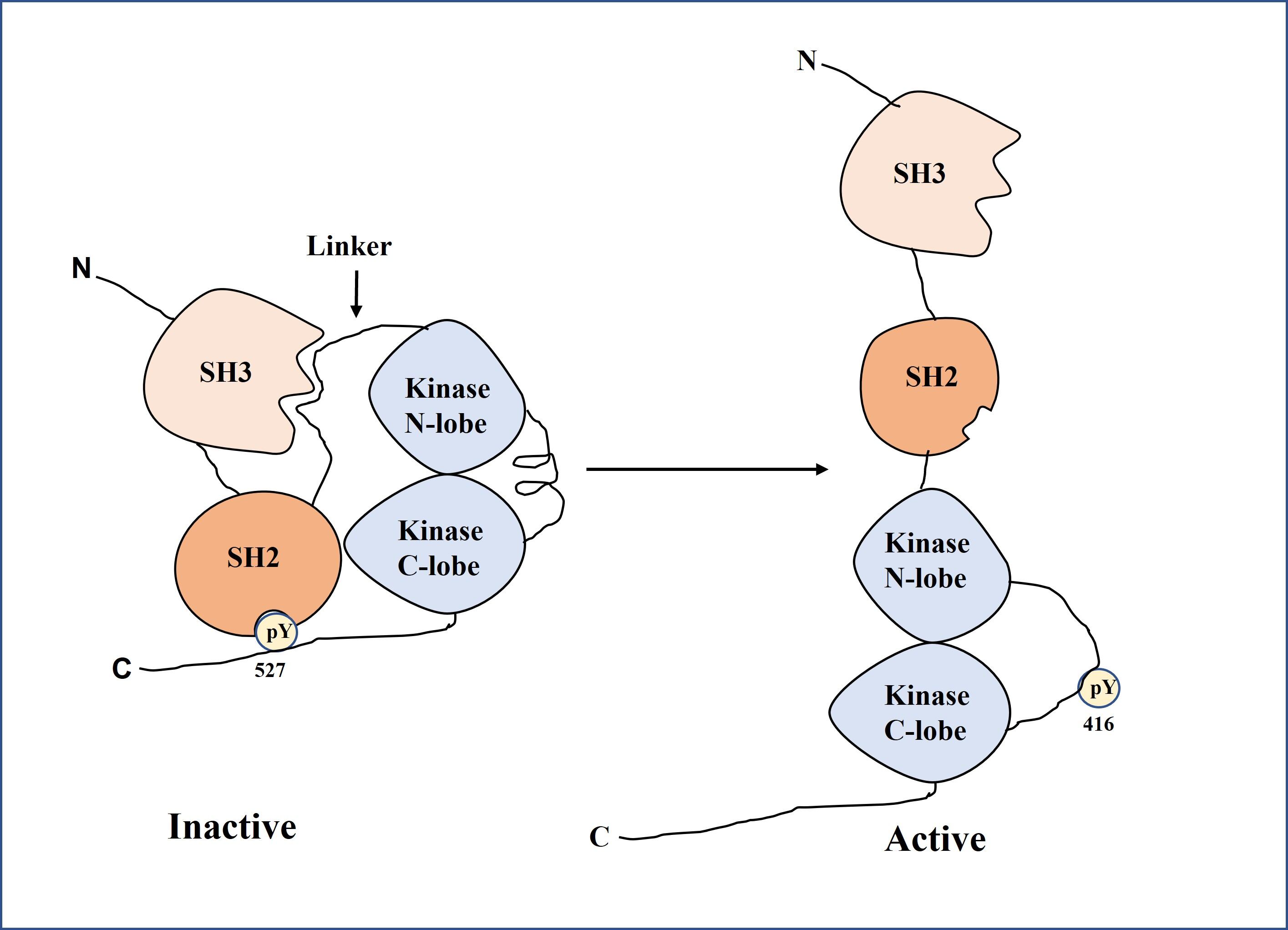
SFKs are structurally related to each other and have structural components that are conserved among family members as shown in Figure 2 which was derived with the modification of literature [10]. These structural components are a short C-terminal tail, an Src module that contains Src Homology-3 domain (SH3), Src Homology-2 domain (SH2), and a tyrosine kinase domain, an N-terminal myristoylation site and a palmitoylation site in most members, which oversee cell localization, and a "unique" domain that differs among family members [11]. The Src module is based on the SH2-kinase linker, which is connected on one face by the usual ligand-binding pocket of the SH3 domain and forms a polyproline type II helix [11]. The kinase cannot be activated because the other face of this linker is jammed against the N-lobe of the kinase domain. An exogenous ligand that displaces the SH3 domain can substantially activate Src-family kinases [11].
Figure 2. Conformational states of Src family kinases (SFKs). In the inactive state (left), SH3 binds the SH2-kinase linker and SH2 engages phosphorylated Tyr527, locking the kinase in a closed form. In the active state (right), ligand binding to SH3, Tyr527 dephosphorylation, or Tyr416 phosphorylation disrupts these interactions, producing an open, catalytically active conformation.
The primary mode of control in Src-family kinases is the interaction of two tyrosine phosphorylation sites: the conserved positive regulatory phosphorylation site in the activation loop and a negative regulatory site on a short tail at the C-terminus of the kinase domain. For example, Tyr-416 in chicken c-Src and Tyr-419 in human c-Src phosphorylation in the activation loop of the kinase domain activates the enzyme; Tyr-527 in chicken c-Src and Tyr-530 in human c-Src phosphorylation in the C-terminal tail inhibits the enzyme activity [8-12]. Tyr-416 or Tyr-527, but not both, is where the enzyme is phosphorylated in vivo [11]. The Src module in the inactive structure is stabilized by docking its tyrosine-phosphorylated tail to the SH2 domain [11]. When SFKs are activated, they induce cell proliferation, differentiation, migration, and carcinogenesis (Figure 3).
A linker is placed on the pocket of the SH3 domain of the Src module comprising the SH2 and SH3 domains [11,12]. The N-lobe of the kinase domain is attached to the linker site, which is not attached to the SH2 domain. The interplay of two tyrosine phosphorylation sites regulates the Src family kinase: a positive regulatory phosphorylation site in the activation loop and a negative regulatory site on a short tail at the kinase domain C-terminus. When the activation loop is in the inhibitor helix form and the C-terminus of the kinase domain is attached to the SH2 domain through the tyrosine phosphorylation site, the Src family kinase is in the inactive form. When the activation loop is extended and the C-terminus is detached from the SH2 domain, the Src family kinase remains active. The Src family kinase cannot be activated unless the N-lobe of the kinase domain is unclogged by the other face of the linker [10]. Indeed, an exogenous ligand that displaces the SH3 domain can substantially activate SFKs by the autophosphorylation of 416Y of the activation loop [12].
2.2. SFKs In Epidermal Maintenance
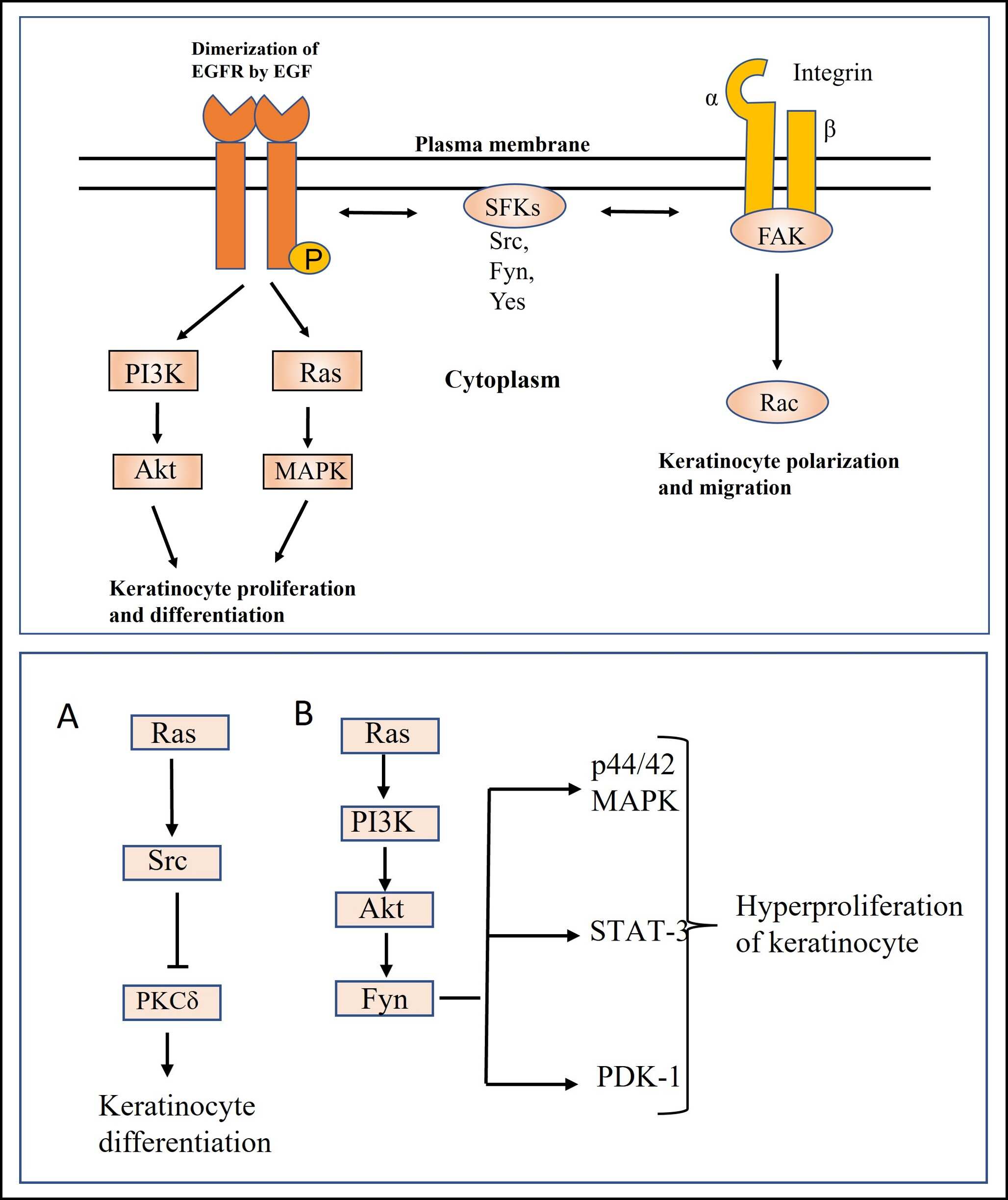
Nonreceptor-type PTKs, known as Src family kinases (SFKs), which include Src, Fyn, and Yes, are crucial for controlling cell proliferation and differentiation, migration, and carcinogenesis[8, 13]. They are believed to activate downstream Ras-MAPK and PI3K pathways by interacting with RPTKs, such as the EGFR expressed in keratinocytes (Figure 3), and are thought to be involved in epidermal cell proliferation and differentiation (Table 1) [14]. The members of the Src and Ras families both promote proliferation, but they induce distinct changes in the terminal differentiation program of keratinocytes. Cells expressing Src exhibit altered morphology and reduced expression of differentiation markers, whereas Ras affects differentiation in a different manner [15]. Src kinase can induce epidermal differentiation-like morphological changes in A431 cells through a novel pathway that is independent of both its SH3 domain and the Ras signaling cascade but potentially through cytoskeletal or adhesion-related proteins [16]. Activation of epidermal growth factor receptor (EGFR) by its ligand phosphorylates the tyrosine residue of the Src-activating and signaling molecule (Srcasm). The phosphorylation of Srcasm activates Src kinase [17]. Calautti et al. reported that the PI3-Akt pathway causes early keratinocyte differentiation through Src tyrosine kinase and epidermal growth factor receptor[18]. A study showed during early differentiation, c-Src was activated, and c-Yes kinase was deactivated in human cultured keratinocytes treated with calcium. c-Src kinase was activated due to the dephosphorylation of tyrosine by phosphatase enzyme [19]. In addition, in a recent study, it has been reported that in young skin yes associated protein YAP with TAZ maintains proliferation of skin stem cells. With aging, YAP/TAZ activity decreases, which results in decreased proliferation of basal stem cells [20]. Vitamin D signaling, which promotes keratinocyte differentiation and barrier function, may intersect with SFK pathways to maintain epidermal integrity because of its broader role in skin health that could complement SFK-mediated processes [21]. Therefore, it is concluded that SFKs are involved in regulating the proliferation and differentiation of keratinocytes, and EGF is a critical driver for keratinocyte survival and proliferation. Src facilitates the adhesion and cellular communication of the epidermal layers.
Table 1. Summary of some literature review about the functional roles and mechanistic pathways of Src family kinases in keratinocyte biology.
Src kinase is activated in keratinocytes during the early stage of wound healing [22]. During this process, integrin α3β1 induces autophosphorylation of FAK at Y397, followed by Src kinase–mediated phosphorylation at Y861 and Y925. This caused Rac1 activation, leading to the polarization and migration of keratinocytes (Figure 3) [23]. Detergent-resistant membranes (DRMs) are a domain of Src kinase that inhibits its function. In wound-induced keratinocytes, type II keratins 6 bind to this domain and inhibit Src kinase, resulting in the inhibition of substrates such as FAKs, P130Cas, and paxillin, which inhibits cell migration [24]. On the other hand, Wu et al. reported that the extracellular signal-regulated kinase (ERK) and c-Jun-N-terminal kinase (JNK) pathways induced Src-mediated increased MMP-2 expression and decreased E-cadherin expression, thereby causing wound healing through migration [6]. This paper also revealed that in vitro, Src overexpression markedly enhanced cell migration, and in vivo, it accelerated wound healing. On the other hand, both processes were delayed by silencing Src [6]. Keratinocyte proliferation was not significantly impacted by Src, indicating that its main function is to promote migration. All the above reports indicate that SFKs play a crucial role in wound healing by triggering keratinocyte migration at the wound site through integrin-mediated activation. Additionally, SFKs facilitate tissue remodeling by the deposition of new extracellular matrix components [6].
2.4. SKFs In Cancer Formation (Tumorigenesis)
The carboxy-terminal Src kinase (CSK) is a PTK that catalyzes the phosphorylation of a COOH-terminal regulating tyrosine residue, thus blocking the activity of all SFKs [13, 25]. Ablation of CSK showed defective cell adhesion in keratinocytes with hyperplasia, improper differentiation, and inflammation due to upregulation of RAC1 activity [26]. The activity and expression of Src family kinases are regulated during the normal hair cycle. Its activity and expression were upregulated in the anagen phase and in the tumor-inducing model [27]. In the tumor-inducing model, the Src inhibitor AZD0530 reduced papilloma formation, but AZD0530 did not inhibit subsequent papilloma proliferation, indicating the involvement of Src in early cancer development [27]. As described in Figure 3, In Ras-transformed mouse keratinocytes, Src caused the inactivation of PKCδ by tyrosine phosphorylation, thereby altering the differentiated phenotype and tight junction formation, which is responsible for cancer formation [28]. In Ras-transformed keratinocytes, Fyn expression was upregulated, and inhibition of FYN using siRNA caused Rho GTPase activation that caused polymerization of F-actin [29]. Fyn is an effector molecule of the Ras-PI3-Akt pathway that causes keratinocyte invasion [30]. In 2007, Li et al. reported that Fyn overexpression causes thickened, hyperplastic, and scaly epidermis through activation of important signaling molecules, including PDK-1, STAT-3, and p44/42 MAP kinases, that support the growth of epithelial cells in mice [31]. Sarcasm can restore the homeostasis of keratinocytes through downregulation of Fyn [30]. A number of distinct tyrosine phosphorylation sites in Sarcasm/Srcasm are phosphorylated by activated SFKs [32]. When SFKs are phosphorylated, Sarcasm/Srcasm binds to them and stops their activity through a lysosomal-dependent mechanism. This works to limit keratinocyte proliferation and promote keratinocyte differentiation by blocking the activation of PDK1/Akt/mTOR, MEK/ERK, and STAT3 [17, 31, 32]. Cell growth, survival, and cell cycle advancement are all aided by the PI3K/AKT/mTOR signaling pathway [33].
On the other hand, the JAK/STAT and Ras/Raf/MEK/ERK pathways induce cell cycle progression and inhibit apoptosis [34]. In cutaneous squamous cell carcinoma (SCC), Srcasm is downregulated, and Fyn activity is increased, which causes downregulation of Notch1 and P53 [35]. In the epidermis, Notch1 acts like a tumor suppressor gene. Notch1 deletion mediated by Cre-recombinase in keratinocytes creates a microenvironment that induces tumor formation [36]. In addition, P53 loss in vitro and in a mouse, model developed skin cancer [37]. On the other hand, extracellular Ca²⁺ ion induced Fyn/Src activity through G-protein coupled receptors that resulted in adherens junction formation in keratinocytes.
Figure 3. The downstream signaling pathways of integrins or growth factor receptors (upper panel) and the function of Src family kinases (SFKs) in controlling keratinocyte proliferation and differentiation (lower panel). EGF, epidermal growth factor; FAK, focal adhesion kinase.
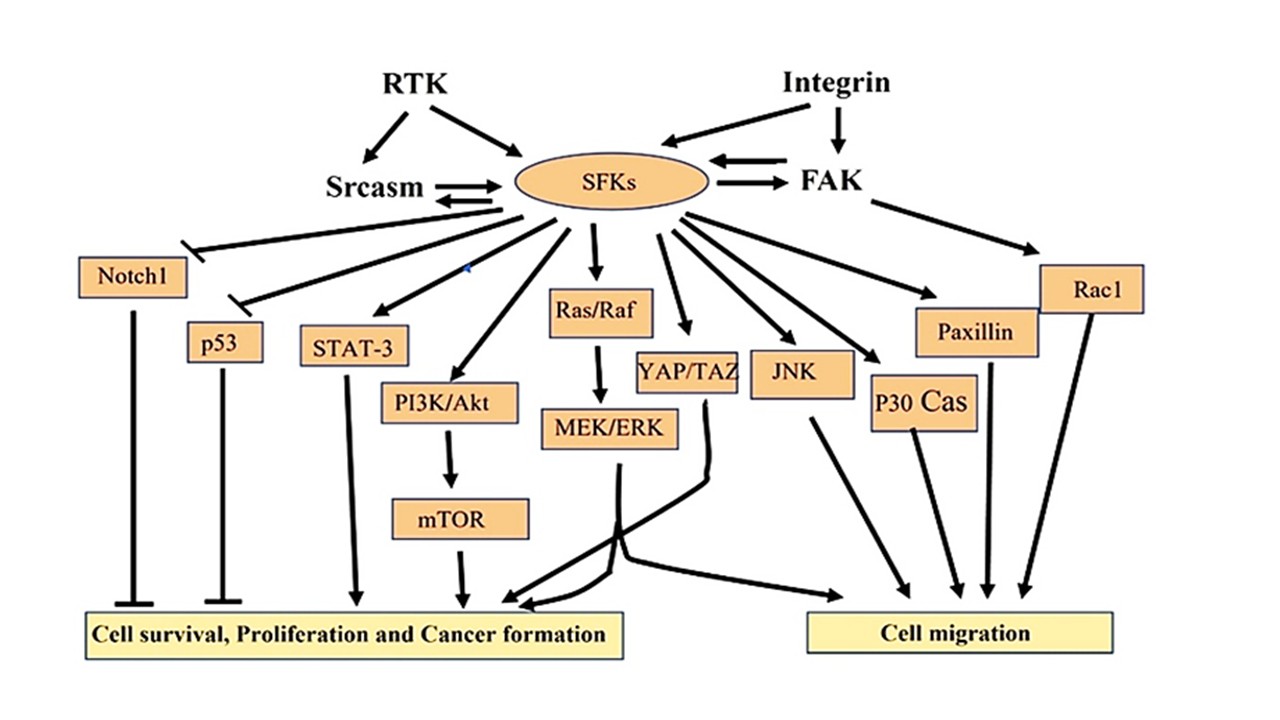
Activation of Fyn/Src increased intracellular Ca2+ ions, thereby resulting in differentiation of keratinocytes [38]. Thus, Fyn shows a contradictory function, resulting in adherens junction formation during keratinocyte differentiation and adherens junction dissolution in keratinocyte transformation and oncogenesis [39]. Montagner et al. reported that peroxisome proliferator-activated receptor (PPAR) β/δ was increased in mice with ultraviolet (UV)-induced skin cancer, which led to epithelial mesenchymal transition (EMT) and ultimately cancer. In terms of mechanism, UV-induced PPARββ/δ leads to Src overexpression, which in turn triggers the EGFR/Erk1/2 signaling pathway and EMT [40]. The review paper by Ortiz et al. reported that SFKs act as adaptor proteins, which cause EMT through cytoskeletal reorganization. The EMT is required for cancer invasion and metastasis [41], which indicates a role of SFKs in cancer progression. Chronic inflammation is the indicator of UV-mediated skin cancer [42]. 4-phenylpyridine (4-PP), a natural product, binds to Src and stabilizes it. The stabilized Src inhibited the phosphorylation of UV-induced EGFR, thus inhibiting inflammation [43], describing a role of Src in inflammation and cancer formation. From the above literature, it can be summarized that in epidermal cancer, overexpression of Src leads to increased keratinocyte proliferation, promoting invasive behavior through cytoskeletal reorganization by bypassing normal growth controls. To understand the role of SFKs in keratinocyte migration and cancer formation, Figure 4 summarizes the mechanisms.
Figure 4. Summary of the mechanism by which SFKs play crucial role in cell survival, proliferation, migration, and cancer formation. This figure was drawn on the basis of reference [8].
The Yes-associated protein YAP is located in basal progenitor cells, promoting their proliferation and preventing their differentiation [44]. The Hippo/YAP signaling pathway regulates cell proliferation and organ size [45, 46]. As an effector of the Hippo signaling pathway, overactive YAP causes liver, lung, and colorectal cancer, and mutations in Hippo pathway components exhibit overgrowth of organs, increased stemness, and decreased cell differentiation [47, 48]. It has recently been reported that YAP, a mechanosensory, is activated due to tissue stiffness regardless of Hippo kinase pathway activation [49, 50]. The above data clearly show that YAP activity must be strictly regulated for epidermal homeostasis and that abnormal nuclear YAP activity leads to epidermal tumor growth. Akladios et al. reported that overexpression of mutant YAP localized in the nucleus causes increased keratinocyte proliferation in mouse skin through activation of β-catenin [51].
They also reported that YAP also causes the overexpression of Gli2 through β-catenin activity that finally results in basal cell carcinoma (BCC) [52]. These results indicate that there is a crosstalk between the YAP and Wnt signaling pathways that drive hyperplasia and BCC.
3. SFKS TARGETING AGENTS IN cSCC DEVELOPMENT
Table 2represents the summary of SFKs targeting agents in cSCC development. The subsequent part discusses the medications and treatments that target SFKs in order to inhibit tumor growth and restore normal tissue architecture. The only FDA-approved SFK inhibitor for treatment of Philadelphia chromosome-positive acute lymphocytic leukemia (ALL) or chronic myeloid leukemia (CML) is dasatinib, also referred to as BMS-354825 [8]. It has been reported that it also acts as an inhibitor of the c-Src and MAPK pathways in the cSCC cell line. This inhibition induced apoptosis of cSCC cells [53]. Topical application of dasatinib also inhibited cSCC development in the transgenic eK14-Fyn Y528F mice skin cancer model [54]. In head and neck squamous cell cancer (HNSCC), which is quite similar to cSCC, bosutinib reduced Src and EGFR phosphorylation. In vivo xenograft studies using cells taken from HNSCC produced comparable outcomes. In both cell lines and xenografts, it inhibited growth and caused apoptosis [55].
Table 2. Summary of Src family kinase (SFK)-targeting agents with potential relevance to cutaneous squamous cell carcinoma (cSCC).
Even though the trials were not conducted in cSCC specifically, the process suggests that they could be applied there. Abl and SFKs are both inhibited by the dual tyrosine kinase inhibitor AZD-0530. For solid tumors, AZD-0530 is being used in several phase II studies [8]. According to reports, it also inhibits keratinocyte migration and proliferation by blocking Src family kinases [27], which could be helpful in cSCC. In addition, pharmaceutical phytochemicals can be utilized to prevent skin disorders because it has been reported that 4-phenylpyridine, isocitric acid, caffeic acid, danshensu etc. regulate SFKs, which are important targets for avoiding environmental stress-induced skin cancer [56].
4. CONCLUSION
Src family kinases (SFKs) are key regulators of epidermal biology, organizing important processes such as keratinocyte proliferation and differentiation. As discussed in this review, SFKs regulate epidermal homeostasis by balancing stem cell renewal and terminal differentiation, thereby preserving the stratum corneum and its barrier function. The dysregulation of SFK activity upsets this balance, contributing to disastrous results. For example, SFK signaling overactivation accelerates wound healing and causes carcinogenesis, notably in SCC. Understanding the roles of SFKs will be helpful for wound care and cancer treatment. Future studies should focus on tissue-specific SFK isoforms and their interactions with immune cells or the extracellular matrix during wound closure and cancer progression. In addition, overexpression of SFKs leads to cancer; however, it is unknown if SFK mutation results in aberrant keratinocyte proliferation. Researchers can investigate whether an SFK mutation can result in cancer. We believe that more research on SFKs in keratinocytes is still possible. Such findings could improve therapeutic techniques, allowing tailored interventions for patients with epidermal disorders.
ACKNOWLEDGEMENT
We extend our heartfelt gratitude to all those who contributed to the successful completion of this research project.
FUNDING SOURCES
This research was conducted with self-funding. Therefore, any kind of financial support was not received for this study.
CONFLICT OF INTEREST
The authors declare no conflict of interest. All aspects of this research were conducted impartially and independently. No financial or personal relationships with other people or organizations have influenced this work.
ETHICS STATEMENT
This study did not involve any experiments on human participants or animals; therefore, formal written informed consent was not required. All figures in this study were created with modifications based on relevant referenced sources; therefore, no permission for reuse is required for any figure presented herein.
References
Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10(3):207-217.
Fuchs E. Scratching the surface of skin development. Nature. 2007;445(7130):834-842.
Eckhart L, Lippens S, Tschachler E, Declercq W. Cell death by cornification. Biochim Biophys Acta Mol Cell Res. 2013;1833(12):3471-3480.
Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6(4):328-340.
Pastushenko I, Prieto-Torres L, Gilaberte Y, Blanpain C. Skin stem cells: at the frontier between the laboratory and clinical practice. Part 1: epidermal stem cells. Actas Dermosifiliogr (Engl Ed). 2015;106(9):725-732.
Wu X, Yang L, Zheng Z, Li Y, Chen Y, Jia Y, Ma J, Jin Q, Cheng B, Zhao J. Src promotes cutaneous wound healing by regulating MMP-2 through the ERK pathway. Int J Mol Med. 2016;37(3):639-648. doi:10.3892/ijmm.2016.2472.
Owens DW, McLean GW, Wyke AW, Paraskeva C, Parkinson EK, Frame MC. The catalytic activity of the Src family kinases is required to disrupt cadherin-dependent cell-cell contacts. Mol Biol Cell. 2000;11(1):51-64. doi:10.1091/mbc.11.1.51.
Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist. 2009;14(7):667-678. doi:10.1634/theoncologist.2009-0009.
Rous P. A transmissible avian neoplasm. (Sarcoma of the common fowl) by Peyton Rous, M.D., Experimental Medicine for Sept. 1, 1910, vol. 12, pp.696-705. J Exp Med. 1979;150(4):738-753. doi:10.1084/jem.150.4.729
Boonyaratanakornkit V, Edwards DP. Receptor mechanisms of rapid extranuclear signalling initiated by steroid hormones. Essays Biochem. 2004; 40:105-120. doi:10.1042/bse0400105.
Shah NH, Amacher JF, Nocka LM, Kuriyan J. The Src module: an ancient scaffold in the evolution of cytoplasmic tyrosine kinases. Crit Rev Biochem Mol Biol. 2018;53(5):535-563. doi:10.1080/10409238.2018.1495173.
Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3(5):629-638. doi:10.1016/s1097-2765(00)80356-1.
Okada M. Regulation of the SRC family kinases by CSK. Int J Biol Sci. 2012;8(10):1385-1397.
Ayli EE, Li W, Brown TT, Witkiewicz A, Elenitsas R, Seykora JT. Activation of Src-family tyrosine kinases in hyperproliferative epidermal disorders. J Cutan Pathol. 2008;35(3):273-277.
Weissman B, Aaronson SA. Members of the src and ras oncogene families supplant the epidermal growth factor requirement of BALB/MK-2 keratinocytes and induce distinct alterations in their terminal differentiation program. Mol Cell Biol. 1985;5(12):3386-3396.
Jin F, Reynolds AB, Hines MD, Jensen PJ, Johnson KR, Wheelock MJ. Src induces morphological changes in A431 cells that resemble epidermal differentiation through an SH3- and Ras-independent pathway. J Cell Sci. 1999;112(17):2913-2924.
Li W, Marshall C, Mei L, Gelfand J, Seykora JT. Srcasm modulates EGF and Src-kinase signaling in keratinocytes. J Biol Chem. 2005;280(7):6036-6046. doi:10.1074/jbc.M406546200.
Calautti E, Li J, Saoncella S, Brissette JL, Goetinck PF. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J Biol Chem. 2005;280(38):32856-32865.
Zhao Y, Sudol M, Hanafusa H, Krueger J. Increased tyrosine kinase activity of c-Src during calcium-induced keratinocyte differentiation. Proc Natl Acad Sci U S A. 1992;89(17):8298-8302.
Kim JY, Quan T. Emerging perspectives of YAP/TAZ in human skin epidermal and dermal aging. Ann Dermatol. 2024;36(3):135-144. doi:10.5021/ad.23.156.
Ali MT, Shipu SJ, Naima NJ, Islam GGMR, Ahmed MS, Alam MA. Vitamin D Status and Knowledge in Relation to Demographic and Lifestyle Factors: A Clinical Data-Based Cross-Sectional Study. J. Biosci. Public Health 2025; 1(2):17-26. doi:10.5455/JBPH.2025.07.
Yamada T, Aoyama Y, Owada MK, Kawakatsu H, Kitajima Y. Scraped-wounding causes activation and association of C-Src tyrosine kinase with microtubules in cultured keratinocytes. Cell Struct Funct. 2000;25(6):351-359.
Choma DP, Milano V, Pumiglia KM, DiPersio CM. Integrin α3β1-dependent activation of FAK/Src regulates Rac1-mediated keratinocyte polarization on laminin-5. J Invest Dermatol. 2007;127(1):31-40.
Rotty JD, Coulombe PA. A wound-induced keratin inhibits Src activity during keratinocyte migration and tissue repair. J Cell Biol. 2012;197(3):381-389.
Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. 1991;351(6321):69-72.
Yagi R, Waguri S, Sumikawa Y, Nada S, Oneyama C, Itami S, Schmedt C, Uchiyama Y, Okada M. C-terminal Src kinase controls development and maintenance of mouse squamous epithelia. EMBO J. 2007;26(5):1234-1244.
Serrels B, Serrels A, Mason SM, Baldeschi C, Ashton GH, Canel M, Byron A, Frame MC, Brunton VG. A novel Src kinase inhibitor reduces tumour formation in a skin carcinogenesis model. Carcinogenesis. 2009;30(2):249-257. doi:10.1093/carcin/bgn278.
Joseloff E, Cataisson C, Aamodt H, Ocheni H, Blumberg PM, Kraker AJ, Yuspa SH. Src family kinases phosphorylate protein kinase C δ on tyrosine residues and modify the neoplastic phenotype of skin keratinocytes. J Biol Chem. 2002;277(14):12318-12323.
Fenton SE, Hutchens KA, Denning MF. Targeting Fyn in Ras-transformed cells induces F-actin to promote adherens junction-mediated cell–cell adhesion. Mol Carcinog. 2015;54(10):1181-1193.
Yadav V, Denning MF. Fyn is induced by Ras/PI3K/Akt signaling and is required for enhanced invasion/migration. Mol Carcinog. 2011;50(5):346-352.
Li W, Marshall C, Mei L, Gelfand J, Seykora JT. Srcasm corrects Fyn-induced epidermal hyperplasia by kinase down-regulation. J Biol Chem. 2007;282(2):1161-1169. doi:10.1074/jbc.M606583200.
Seykora JT, Mei L, Dotto GP, Stein PL. Srcasm: a novel Src activating and signaling molecule. J Biol Chem. 2002;277(4):2812-2822. doi:10.1074/jbc.M106813200.
Glaviano A, Foo ASC, Lam HY, Chuah SW, Lee M, Aung KT, Ong SM, Li L, Ng LG, Chen Q, Thike AA, Tan PH, Lim B, Bay BH. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer. 2023;22(1):138. doi:10.1186/s12943-023-01827-6.
Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18(2):189-218. doi: 10.1038/sj.leu.2403241.
Zhao L, Li W, Marshall C, Griffin T, Hanson M, Hick R, Seykora JT. Srcasm inhibits Fyn-induced cutaneous carcinogenesis with modulation of Notch1 and p53. Cancer Res. 2009;69(24):9439-9447.
Demehri S, Turkoz A, Kopan R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell. 2009;16(1):55-66. doi: 10.1016/j.ccr.2009.05.016.
Page A, Navarro M, Suarez-Cabrera C, Pizarro J, Santos M, Paramio JM, Jorcano JL. Protective role of p53 in skin cancer: Carcinogenesis studies in mice lacking epidermal p53. Oncotarget. 2016;7(15):20902-20918. doi:10.18632/oncotarget.7897.
Bikle DD, Xie Z, Tu CL. Calcium regulation of keratinocyte differentiation. Expert Rev Endocrinol Metab. 2012;7(4):461-472. doi:10.1586/eem.12.34.
Fenton SE, Denning MF. FYN signaling: divergent adhesive functions for Fyn in keratinocytes. Exp Dermatol. 2015;24(2):81-85.
Montagner A, Delgado MB, Tallichet-Blanc C, Chan JS, Sng MK, Mottaz H, Degrelle SA, Huber C, Wahli W, Tan NS, Michalik L, Desvergne B. Src is activated by the nuclear receptor peroxisome proliferator-activated receptor β/δ in ultraviolet radiation-induced skin cancer. EMBO Mol Med. 2014;6(1):80-98. doi:10.1002/emmm.201302666.
Ortiz MA, Mikhailova T, Li X, Porter BA, Bah A, Kotula L. Src family kinases, adaptor proteins and the actin cytoskeleton in epithelial-to-mesenchymal transition. Cell Commun Signal. 2021;19(1):67. doi:10.1186/s12964-021-00750-x.
Maru GB, Gandhi K, Ramchandani A, Kumar G. The role of inflammation in skin cancer. Adv Exp Med Biol. 2014; 816:437-469. doi:10.1007/978-3-0348-0837-8_17.
Kim JG, Kang HY, Kim MJ, Kang JS, Lee WJ, Song HS, Lee SJ, Park YH, Park KC, Park KC. 4-phenylpyridine suppresses UVB-induced skin inflammation by targeting c-Src in vitro and in vivo. J Cell Mol Med. 2022;26(14):3891-3901. doi:10.1111/jcmm.17422.
Zhang H, Pasolli HA, Fuchs E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci U S A. 2011;108(6):2270-2275.
Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120-1133. doi: 10.1016/j.cell.2007.07.019.
Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962-1971. doi:10.1101/gad.1664408
Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13(4):246-257. doi:10.1038/nrc3458.
Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol Rev. 2014;94(4):1287-1312. doi:10.1152/physrev.00005.2014.
Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154(5):1047-1059. doi: 10.1016/j.cell.2013.07.042.
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179-183. doi:10.1038/nature10137.
Akladios B, Mendoza-Reinoso V, Samuel MS, Hardeman EC, Khosrotehrani K, Key B, Hogan BM, Lawlor KE, Johnston ST, Henderson MA, Dworkin S, Ernst M, Pearson RB, Humbert PO. Epidermal YAP2-5SA-ΔC drives β-catenin activation to promote keratinocyte proliferation in mouse skin in vivo. J Invest Dermatol. 2017;137(3):716-726. doi: 10.1016/j.jid.2016.10.029.
Akladios B, Mendoza Reinoso V, Cain JE, Khosrotehrani K, Pearson HB, Pearson RB, Humbert PO. Positive regulatory interactions between YAP and Hedgehog signalling in skin homeostasis and BCC development in mouse skin in vivo. PLoS One. 2017;12(8):e0183178. doi: 10.1371/journal.pone.0183178.
Farshchian M, Nissinen L, Grénman R, Kähäri VM. Dasatinib promotes apoptosis of cutaneous squamous carcinoma cells by regulating activation of ERK1/2. Exp Dermatol. 2017;26(1):89-92. doi:10.1111/exd.13109.
Segrelles C, Contreras D, Navarro EM, Gutiérrez-González A, García-Escudero R, Garín MI, Paramio JM, Lorz C. Bosutinib inhibits EGFR activation in head and neck cancer. Int J Mol Sci. 2018;19(7):1824. doi:10.3390/ijms19071824.
Paik SJ, Kim DJ, Jung SK. Preventive effect of pharmaceutical phytochemicals targeting the Src family of protein tyrosine kinases and aryl hydrocarbon receptor on environmental stress-induced skin disease. Int J Mol Sci. 2023;24(6):5953. doi:10.3390/ijms24065953.
 access
access