J. Biosci. Public Health. 2025; 1(3)
Corresponding Author *
![]() https://orcid.org/0009-0007-3167-0903
https://orcid.org/0009-0007-3167-0903
Affiliations:
Department of Biochemistry and Molecular Biology, Gono University, Dhaka 1344, Bangladesh.
Corresponding Author *
![]() https://orcid.org/0009-0007-3167-0903
https://orcid.org/0009-0007-3167-0903
Affiliations:
Department of Biochemistry and Molecular Biology, Gono University, Dhaka 1344, Bangladesh.
Coauthor
mdrakibulhasanbmb13.gb@gmail.com
![]() https://orcid.org/0009-0007-4355-0327
https://orcid.org/0009-0007-4355-0327
Affiliations:
Department of Biochemistry and Molecular Biology, Gono University, Dhaka 1344, Bangladesh.
Coauthor
![]() https://orcid.org/0009-0006-0573-6473
https://orcid.org/0009-0006-0573-6473
Affiliations:
Department of Biochemistry and Chemistry, Sylhet Agricultural University, Sylhet 3100, Bangladesh.
Coauthor
Affiliations:
https://orcid.org/0009-0008-4318-7772
Coauthor
Affiliations:
https://orcid.org/0009-0003-6692-2577
Coauthor
![]() Department of Biochemistry and Chemistry, Sylhet Agricultural University, Sylhet 3100, Bangladesh.
Department of Biochemistry and Chemistry, Sylhet Agricultural University, Sylhet 3100, Bangladesh.
Affiliations:
https://orcid.org/0009-0006-4630-8942
Coauthor
![]() Department of Biochemistry and Chemistry, Sylhet Agricultural University, Sylhet 3100, Bangladesh.
Department of Biochemistry and Chemistry, Sylhet Agricultural University, Sylhet 3100, Bangladesh.
Affiliations:
https://orcid.org/0000-0001-9347-5811
The structural and functional investigation of a hypothetical protein HP (KZV07215.1) from Saccharomyces cerevisiae (SC) has shed light on how fungal adaptation to unusual conditions and metabolic modifications occur. In this study, we performed a comprehensive in silico characterization of a mitochondrial hypothetical protein (KZV07215.1), encoded by the WN66_06778 gene, to elucidate its putative structure and function. Physicochemical profiling revealed a moderately acidic, hydrophobic, and highly stable 58.7-kDa protein with an isoelectric point of 6.55. According to the results of functional annotating programs like InterProScan, NCBI-CDD Search, and Pfam, the desired product is a cytochrome c oxidase (COX) protein of the respiratory system. Its 3D configuration was determined using the homology modeling technique by I-TASSER (TM-score estimation of 0.98 ± 0.05). Following the GalaxyRefine, a stable 3D structure was confirmed by PROCHECK, QMEAN (value of -3.36), QMEANDisCo Global (score of 0.77±0.05), and ERRAT (score of 92.381). FTSite confirms the site of activity of the hypothesized structure. Molecular docking revealed substantial binding affinities with HEA compounds, yielding docking scores of −159.90 kcal/mol (HP-HEA III), ensuring robust and stable interactions. these findings strongly support the annotation of KZV07215.1 as a COX-I-like protein involved in electron transport chain complex IV activity. This work highlights the value of computational pipelines for functional annotation of uncharacterized mitochondrial proteins and suggests potential implications for understanding respiratory regulation and bioenergetic disorders.
Saccharomyces cerevisiae (SC) is a is a unicellular, yellow-green, spherical yeast belonging to the fungi kingdom, having 16 chromosomes and a total genomic DNA of 12068 kilobases (kb). It is a tiny, single-cell fungus that can be readily cultured. It takes quick production, around 1.25-2 hours, to double at a temperature of 30°C, and allows the affordable maintenance of different variants [1]. As a facultative anaerobe, SC survives in both oxygen-rich and oxygen-limited environments, deriving energy through mitochondrial oxidative phosphorylation and the electron transport chain, as well as through fermentative pathways. Because of its metabolic versatility and genetic tractability, SC has long served as a cornerstone in mitochondrial genetics. Pioneering studies on respiratory-deficient “petite” mutants established cytoplasmic inheritance [2], and SC became the organism in which mitochondrial genes [3] and their mosaic intronic architecture [4, 5] were first described. It was also the source of the earliest sequenced mitochondrial gene [6, 7]. Publication of the complete yeast genome in 1996 marked a turning point in functional genomics, revealing that nearly half (56%) of annotated genes lacked experimentally defined roles despite decades of biochemical and genetic research [8-11]. This unexpected knowledge gap stimulated large-scale functional genomics initiatives aimed at characterizing previously unknown proteins [12]. Even today, more than half of the predicted proteins in S. cerevisiae remain functionally uncharacterized, reflecting substantial gaps in our understanding of mitochondrial biology and proteome complexity. Experimental elucidation of such proteins is challenging due to technical, temporal, and financial constraints. Consequently, integrative computational approaches provide a powerful and efficient alternative for predicting the structural and functional properties of hypothetical mitochondrial proteins [13].
The respiratory chain’s terminal enzyme, cytochrome C oxidase (COX), catalyzes the coordinated movement of ferrocytochrome C's multiple (four) electrons towards oxygen molecules. Again, pumping protons also moves from the mitochondrial matrix to the mitochondrial inner cytosolic portion [14]. The catalytically active holoenzyme is made up of 4 reactive metallic cores (Heme A, Heme A3, CuA, and CuB) comprised within a multi-subunit complex. The core of the binuclear reaction is made up of a bridge in-between the completely oxidative relaxation stage of CuB and Heme A3 [15]. Many polypeptide subunits make up the protein matrix that envelops the metal centers. The initial subunit binds the reductive cores of the proteins Heme A, Heme A3, and CuB in COXs originate from prokaryotic systems along with eukaryotic cell types. In addition to binding CuA, subunit II also engages in the binding of cytochrome c, most likely at a location near CuA [16]. The functioning of components I and II's proton transport may be modulated by subunit III [17]. The subunits that are responsible for the making up an active COX molecule are substantially more than three in eukaryotes. The mitochondrial genome codes for the three main subunits (I-III) of the eukaryotic COX complex. They possess basic sequence homology to those of three subunits of bacterial cytochrome c oxidases [18]. The remaining subunits are all products of the nuclear genome. SC serves as a versatile eukaryotic model for researchers to investigate mitochondrial biogenesis and the assembly of respiratory complexes. It can endure the absence of oxidative phosphorylation by using fermentation, which distinguishes it from most organisms. This enables researchers to examine alterations that would be lethal in other creatures. Furthermore, both its nuclear and mitochondrial genomes are amenable to genetic modification, making it an excellent model for investigating mitochondrial gene function [19, 20].
This work aims to define a potential mitochondrial protein of S. cerevisiae (accession no. KZV07215.1) using a comprehensive in silico workflow integrating physicochemical profiling, domain annotation, evolutionary analysis, secondary and tertiary structure prediction, and molecular docking. By elucidating its putative relationship to COX-I, this work provides insights into its potential functional role in the yeast electron transport chain and lays the foundation for future experimental validation.
2.1. Sequence Retrieval
The NCBI (accessed February 2025) (National Center for Biotechnology Information) Protein database was searched for the keyword "Hypothetical proteins AND mitochondrion AND Saccharomyces cerevisiae," and the 766 residues of HP of SC were retrieved. From the list of hits, we randomly selected a hypothetical protein with accession number KZV07215.1 (GI|1023939894|) and retrieved its FASTA sequence for further investigation. A peptide search based on sequence was also carried out to ascertain whether the protein is unnecessary or not, using UniProt peptide search [21].
2.2. Physicochemical Properties Analysis
The ProtParam software in the online ExPASy server (accessed February 2025) was employed in order to evaluate preferred HPs chemically mediated as well as physical features. The analyzer yields theoretical measurements including weight of molecule, amino acid composition, theoretical pI value, count of both positive and negative residues, extinction coefficient, GRAVY score (grand average of hydropathicity), etc [22].
2.3. Functional Domain Annotation
The HP was functionally annotated to make its functions clear. Several freely accessible databases and software, including the NCBI conserved domains database (CDD v3.21, updated 2024) [23], InterProScan v5.57-90.0 (2024 release) [24], and SUPERFAMILY v1.75 [25], were implemented for proper screening of HP’s functional domain and conserver. In every situation, the default parameters were used. The proteins are categorized using the screened-out domains found in the HP. The domains are found using these databases and additional bioinformatics techniques [26].
2.4. Phylogenetic Analysis and Visualization
The UniProtKB (accessed February 2025) [27] was implemented, looking for similar sequences with the examined HP. Using UniProt’s BLAST technique [28]. We looked for similarities in a database of unique proteins. Initially, a total of 42 reviewed homologous protein sequences were identified from the UniProt database. Among them, the first 24 reviewed sequences were downloaded. They were all thought to serve the same function.
The multiple sequence alignment (MSA) was then done by utilizing the MAFFT v7.526 (April 2024) tool [29]. Using homologous sequence alignment, a phylogenetic tree was generated with PhyML v3.0 (release 2024) to highlight the evolutionary segregation of the related proteins [30]. The tree was shown by using the iTOL v6 (accessed February 2025) server [31].
2.5. Secondary Structure Prediction
The prediction of the two-dimensional (2D) structure of HP was shown by the SOPMA servers [32, 33] and PSIPRED v4.0 (2024 release) [34, 35]. With the use of "DATABASE.DSSP," SOPMA makes secondary structure predictions for proteins, whereas PSIPRED utilizes the PSI-BLAST algorithm with feed-forward neural networks [34, 35]. In both cases, the prediction of the 2D structure was performed using the FASTA sequence of HP.
2.6. Tertiary Structure Prediction
The prediction of three-dimensional (3D) structure was carried out by employing the I-TASSER (version 5.1, 2023) servers [36–38]. For homology modeling, we used the default values for all the variables of I-TASSER [39]. The anticipated three-dimensional structures of the HP were improved, and then the YASARA v25.9.17 energy minimization server was implemented to reduce their energy [40]. After that, the enhanced 3D structures were further improved using GalaxyRefine [41]. GalaxyRefine produces several potential structures; the highest-performing and highest-quality ones are hand-selected. The structures of the HP were then converted into three-dimensional forms using PyMOL v2.5.2 [42, 43].
2.7. Model Quality Assessment
SAVES v6.0 (2024 release; accessed March 2025) server’s ERRAT [44] modules along with PROCHECK [45] were accessible for the assessment of HP’s energy-minimization and ideal 3D structure. The 3D model of HP was validated further with the SWISS-MODEL (release 2024) Structural Assessment Tool, which was created by the Swiss Institute of Bioinformatics (SIB) [46, 47]. Finally, the highest standard model was chosen to support further investigation.
2.8. Subcellular Localization Prediction
The optimal functioning of peptides is influenced by their spatial surroundings, which control their biological networks and patterns of interaction [48]. In this particular context, several servers anticipated the HP's subcellular location, including Euk-mPLoc v2.0 [49], TargetP-v2.0 [50], and MitPred v2.0 (accessed March 2025) servers [51].
2.9. Binding Site Prediction and Molecular Docking
The FTSite (2012; accessed March 2025) server was chosen for the determination of the active site and residues of the HP [52]. Quick residue-level annotations are achievable only when the structure and sequence of a protein have a relationship [53]. To ascertain the binding potency of the HP with Heme-A (HEA), molecular docking was conducted utilizing the HDOCK (2024; accessed April 2025) online service [54, 55]. The ligand HEA’s PDB files were acquired from the RCSB PDB [56], and the I-TASSER-generated HP was used as the receptor. The sets of complexes with high negative docking values were selected and retrieved, indicating excellent docking. Version v2.5.2 of PyMOL was used to visualize the obtained result from the HDOCK server [42, 43].
3.1. Retrieval of HP Sequence
The protein database at NCBI was randomly queried, resulting in the HP WN66 06778, which is SC’s HP. Once this sequence was received, UniProt was searched. The HP's properties have been recorded for investigation. Different parameters, which represent the properties of the HP as well as HP’s sequence in FASTA format, have been saved. In this HP of SC, the number of total amino acids is 534, designated as WN66 06778. Other parameters, such as locus, were assigned as KZV07215, likewise accession as KZV07215, and version numbers as KZV07215.1 (Table 1).
Table 1. Representation of distinctive attributes of HP derived from the NCBI protein database.
| Properties | Hypothetical Protein |
| Definition | Hypothetical protein WN66_06778 (mitochondrion) [Saccharomyces cerevisiae] |
| Accession | KZV07215 |
| Version | KZV07215.1 |
| Amino acid | 534 |
| Organism | Saccharomyces cerevisiae |
| FASTA sequence | >KZV07215.1 hypothetical protein WN66_06778 (mitochondrion) [Saccharomyces cerevisiae] MVQRWLYSTNAKDIAVLYFMLAIFSGMAGTAMSLIIRLELAAPGSQYLHGNSQLFNVLVVGHAVLMIFFLVMPALIGGFGNYLLPLMIGATDTAFPRINNIAFWVLPMGLVCLVTSTLVESGAGTGWTVYPPLSSIQAHSGPSVDLAIFALHLTSISSLLGAINFIVTTLNMRTNGMTMHKLPLFVWSIFITAFLLLLSLPVLSAGITMLLLDRNFNTSFFEVAGGGDPILYEHLFWFFGHPEVYILIIPGFGIISHVVSTYSKKPVFGEISMVYAMASIGLLGFLVWSHHMYIVGLDADTRAYFTSATMIIAIPTGIKIFSWLATVYGGSIRLATPMLYAIAFLFLFTMGGLTGVALANASLDVAFHDTYYVVGHFHYVLSMGAIFSLFAGYYYWSPQILGLNYNEKLAQIQFWLIFIGANVIFFPMHFLGINGMPRRIPDYPDAFAGWNYVASIGSFIATLSLFLFIYILYDQLVNGLNNKVNNKSVIYAKAPDFVESNTIFNLNTVKSSSIEFLLTSPPAVHSFNTPAVQS |
NCBI (National Center for Biotechnology Information); HP (hypothetical protein).
3.2. Physicochemical Properties Characterization
The physicochemical characteristics of the selected hypothetical protein (HP) were evaluated using the ExPASy ProtParam server, and the results are summarized in Table 2. The HP consists of 534 amino acids with an estimated molecular weight of 63,295.44 Da. The theoretical isoelectric point (pI) was calculated to be 6.55, indicating that the protein is moderately acidic while retaining a substantial net positive charge at physiological pH. Protein stability is a critical determinant of its structural integrity and functional performance across biological systems. The instability index (II), a commonly used indicator of in vivo stability, predicts a protein to be stable when the score is below 40 [57]. The aliphatic index (AI), which reflects the relative volume of aliphatic side chains and correlates with thermal stability, was calculated as 114.51 showing the target protein's increased thermal stability [58]. The hydropathy value of every amino acid regarding the desired sequence is calculated, and after dividing this value by the total number, we can finally obtain the GRAVY value for the queued protein. When the value of GRAVY is negative, the protein is considered hydrophilic. But in terms of a hydrophobic protein, this value is found as positive. HP has a calculated value of 0.76, indicating that it is a hydrophobic protein. The extinction coefficient, according to the Beer-Lambert rule, measures as a proportionality constant that estimates the power of specific light that is captured by a protein [59]. It was therefore determined that the HP's extinction coefficient was 92250. The high extinction coefficient means that there is an abundance of tyrosine, tryptophan, and cysteine in the surroundings [57]. However, Table 2 represents some other properties of the HP’s physicochemical features.
Table 2. Representation of physicochemical characteristics of HP obtained from the ProtParam tool.
| Parameters | Values |
| Total Number of Negatively Charged Residues (Asp + Glu) | 22
|
| Total Number of Positively Charged Residues (Arg + Lys) | 19 |
| Formula | C2779H4194N646O712S22 |
| Total Number of Atoms | 8353 |
| Estimated Half-Life | 30 hours (mammalian reticulocytes, in vitro) |
| >20 hours (yeast, in vivo) | |
| >10 hours (Escherichia coli, in vivo) |
HP (hypothetical protein), pI (isoelectric point).
3.3. Annotation of Functional Domain
Multiple servers confirmed that the HP contains a well-established conserved domain named Cytochrome c oxidase-like subunit I (COX-I) (Supplementary Table 1). Through asymmetric proton absorption and proton pumping, electrons from COX transfer to oxygen molecules to absorb energy from the cell's membrane potential. In SC, the complex is made up of 11 subunits. All respiring species share an elevated level of preservation in COX-I, one of the main subunits that is encoded within mitochondria. The fundamental protein COX-I is inserted with the redox cofactors copper and heme. Since COX enters the procedure of the assembly process from both sides of the membrane that covers the mitochondria, a high number of assembly components are needed for the enzyme to be assembled. Among the most precisely defined and most preserved assembly-related indicators, Shy1 (SURF-I homologue of yeast) is lost in SC. This causes a developmental deficit in non-biodegradable forms of carbon and a substantial decline in cytochrome oxidase [60]. The hypothesis that Shy1 participates in the Heme A3 insertion phase is supported by recent research. Translational activators for SC COX-I mRNA include Pet309 and the mitochondrial splicing suppressor protein 51 (Mss51). Pet309, the COX-I mRNA activator, belongs to the protein family known as pentatricopeptide repeats (PPR), which is frequently linked to RNA metabolism [61]. The versatile pioneer protein Mss51, the second COX-I-specific activator, lacks any functional domains or patterns in its sequence. Several computational techniques have proposed that HP's unique domain serves as subunit I of cytochrome c oxidase.
3.4. Multiple Sequence Alignment and Analysis of Phylogeny
For conducting MSA (Multiple Sequence Alignment), the UniProt database acted like a BLAST server, returning the values of HP for proteins discovered. This tool was used to search the protein database for microorganisms having the greatest proportion of sequence similarity, the protein and organism name, and the length of amino acid sequences (Supplementary Table 2). Following that, the MAFFT tool was applied to perform sequence alignment. The phylogeny analysis was performed so that it could identify the evolutionary relationship of KZV07215.1 and its origin. The phylogenetic analysis was performed for the first 24 reviewed homologous protein sequences from different organisms. It was observed that the sequences of the HP and Saccharomyces COX-I are 99.3% similar, putting them in a similar clade of the neighbor-joining tree (Figure 1). Nevertheless, the tree also showed us that the COX-I protein of Saccharomyces paradoxus, Candida glabrata, and Kluyveromyces lactis has maximum sequence similarity with that of HP, 99.1%, 91.4%, and 91.4%, respectively. Moreover, Wickerhamomyces canadensis and Vanderwaltozyma polyspora COX-I proteins showed more than 80% sequence similarity with HP of SC (83.9% and 86.3%, respectively).
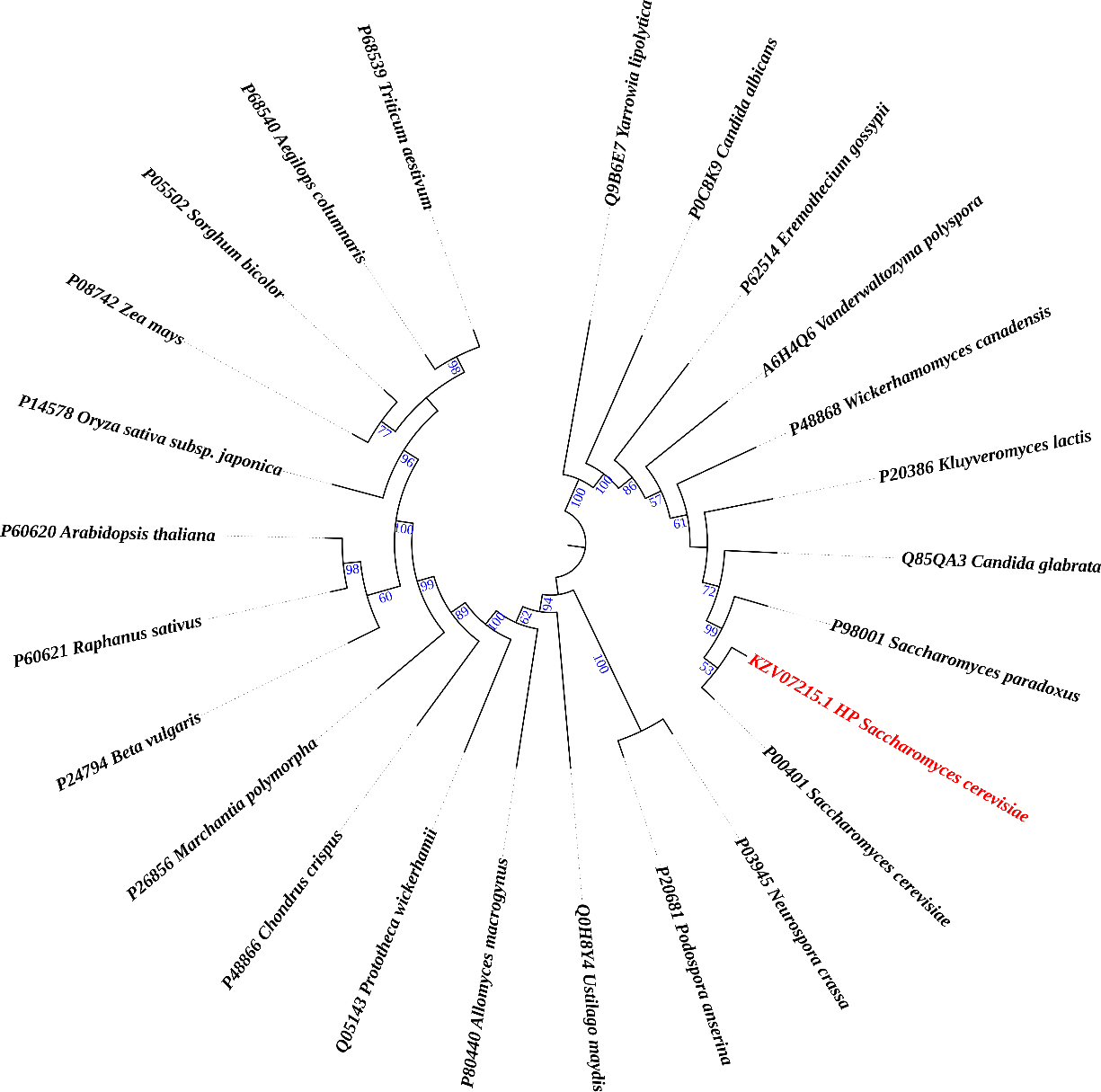
Figure 1. Phylogenetic tree revealing the ancestral relationship of the queued protein (marked as red) with other subunit I of COX proteins.
3.5. Secondary Structure Prediction
The 2D structure prediction was anticipated using a couple of techniques, such as PSIPRED and the SOPMA website. To summarize, the PSIPRED server revealed that our predicted protein structure will have its largest alpha-helix area, followed by random coils, and lastly extended strands (Figure 2). As predicted by PSIPRED, the SOPMA server concurred with it, stating that this HP might have contained a higher fraction of alpha-helix than prolonged strand or random coils (Supplementary Table 3).

Figure 2. Prediction of 2D design of HP conducted by PSI-PRED server. The alpha helix, random coil, and extended strand structures are pointed by pink, ash and yellow colors.
3.6. Tertiary Structure Prediction
The I-TASSER platform was used to precisely anticipate HP models. I-TASSER generated a model for our target protein, selecting one with a TM-score of 0.98±0.05 and a calculated RMSD of 3.6±2.5 from all models produced (Figure 3). A TM value exceeding 0.5 indicates a structure with suitable topology, whereas a TM value below 0.17 suggests random similarity. Consequently, the YASARA server has increased the energy of the protein structure, while the GalaxyRefine server has enhanced the accuracy of its projected models. The program PyMOL was used to analyze and display the three-dimensional structures of the proposed and modified models. The YASARA energy-minimization server reduced the protein structural energy from -219693.9 to -247734.9 kJ/mol. The initial value was -2.59; however, after optimizing the energy of the HP, the final value was -0.48, indicating a comparatively more stable structure.
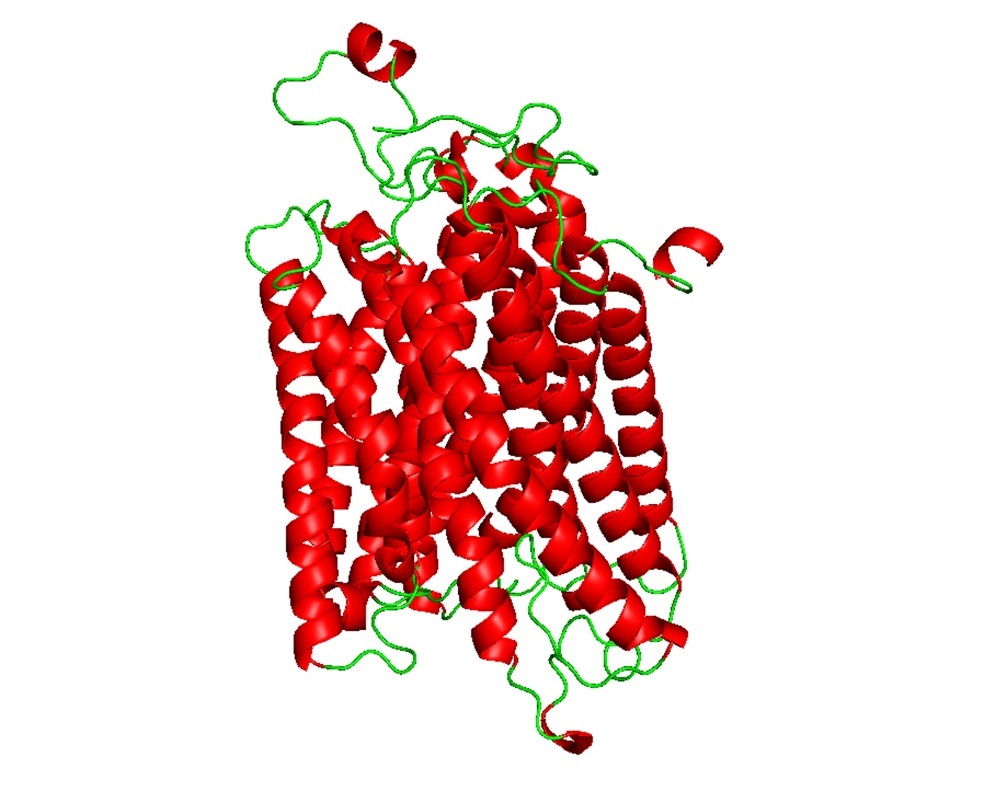
Figure 3. Predicted 3D configuration of the hypothetical protein via I-TASSER server after minimizing energy using YASARA and GalaxyRefine (visualized by PyMOL). The red-spiral represents alpha helices and no beta-sheet arrangements, while the horizontal ribbon stands for the HP's coil structure, in that order. HP stands for hypothetical protein.
3.7. Assessment of Structure Quality
According to the SAVES PROCHECK, 95.5% of the residues of amino acids were situated among a Ramachandran's top choice areas, whereas 3.7% were positioned in additional authorized regions (Figure 4). The ERRAT score of the I-TASSER predicted model is 92.381. According to the SWISS-MODEL, MolProbity gives I-TASSER's HP a value of 1.52, Ramachandran favored region of 97.18%, a QMEAN value of -3.36, and a QMEANDisCo Global of 0.77±0.05 (Supplementary Figure 1). We examined the structure and decided on the I-TASSER model for further investigation. According to ProSA, the predicted Z-score of the HP model is given a negative figure by I-TASSER: -5.71 (Supplementary Figure 2).
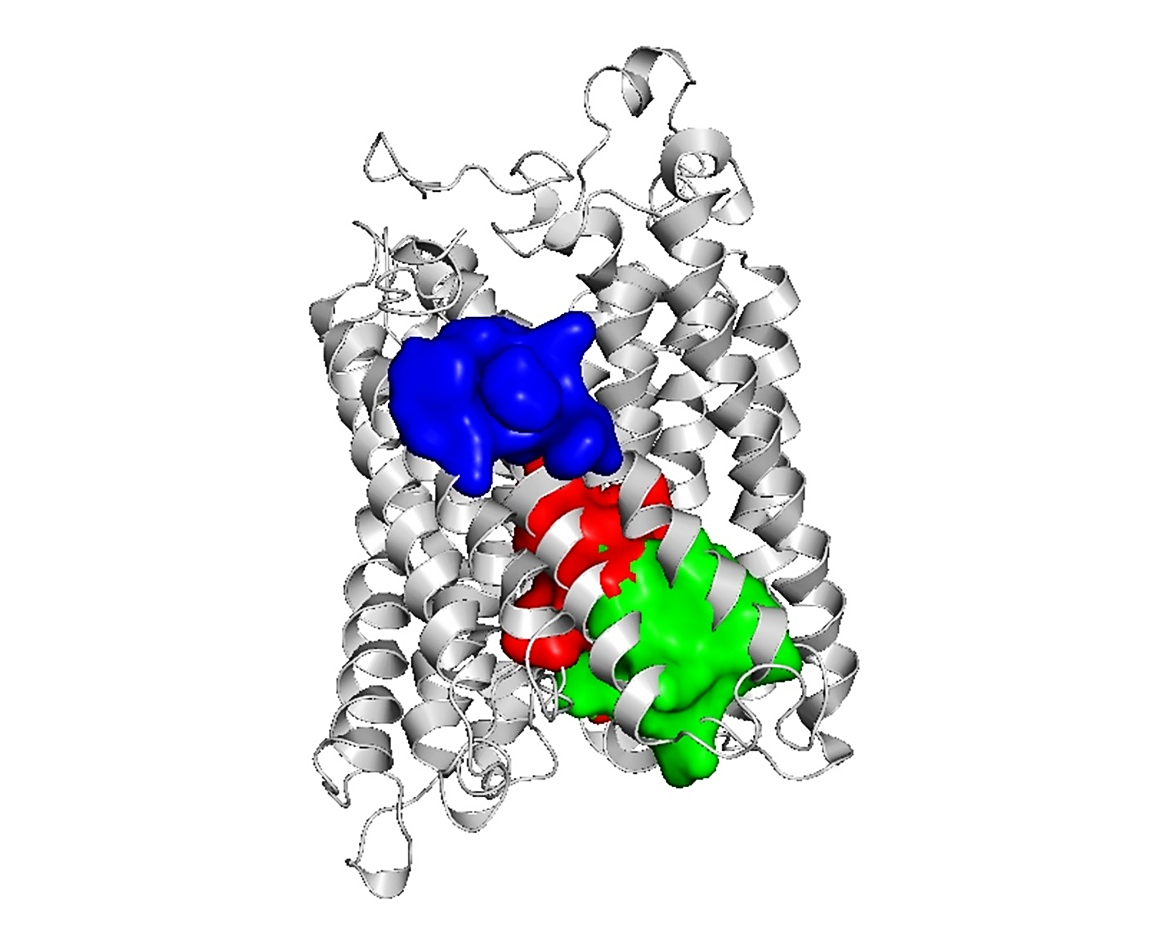
Figure 4. The model displayed through the I-TASSER server's Ramachandran plot. The beta-sheet area and other tertiary structural elements of the HP are represented by the first quadrant, where the alpha-helix area of the left and right hands is represented by the second and third quadrants, respectively. Furthermore, the residues in the most preferred, additional allowed, generously allowed, and prohibited regions are represented by the color regions in red, yellow, gray, and white, respectively.
3.8. Prediction of Subcellular Localization
Several servers, including Euk-mPLoc 2.0, TargetP-2.0, and MitPred, have predicted where the HP will be located within the cell. Various biological activities are connected to a variety of cellular sites [62]. Identifying HP’s location within the cell may shed light on the protein’s probable function. Also, this information could be used to develop a medicine that suppresses the action of the protein being investigated [62]. The researchers proposed that HP belongs to the mitochondrial protein (Supplementary Table 4).
3.9. Prediction of Binding Site
The model's binding site was evaluated using the FTSite server. The amino acid residues inside the binding area were also considered. The results were then shown using the program PyMOL (Figure 5). Characterizing the residues of the binding site is crucial for understanding the functionality of such sites. The active residues of the three main active areas of the model protein are identified using the FTSite estimation (Supplementary Table 5).
Figure 5. Representation of activation sites along with active amino acid molecules predicted by the FTase server. The protein is shown by the cyan hue; on the other hand, the red, green, and blue colors indicate the active sites I, II, and III, respectively.
Upon evaluating the active sites of the target protein using the FTSite server, around thirty amino acid residues were identified. Meanwhile, its active site had five functional amino acid residues, as identified by InterProScan: His-62, His-378, Ser-382, Ile-424, and Ala-461. Based on predictions from InterProScan, NCBI-CD Search, and Pfam, these residues are situated within the COX-I domains of the potential target protein. The HDOCK server was used to analyze the docking patterns of the ligand and the target protein. The target protein (WN66_06778) was conjugated with the ligand Heme A (HEA). A greater negative docking score suggests a more plausible binding model. Our investigation indicates that HP-HEA III has the greatest potential to establish a binding model, with a maximum docking score of -159.90. The probability of the two molecules joining is maximized when the confidence level exceeds 0.7. The likelihood of the two molecules joining is moderate to high when the confidence level ranges from 0.5 to 0.7 and minimal when the value falls below 0.5. The confidence score of HP-HEA III in this instance is 0.5493, indicating a substantial probability of molecular binding. HP-HEA II has a confidence score of roughly 0.5, indicating its propensity to bind. Generally, a closer alignment of the docking posture with the ligand's binding mode results in a reduced root mean square deviation (RMSD) seen throughout the experiment.
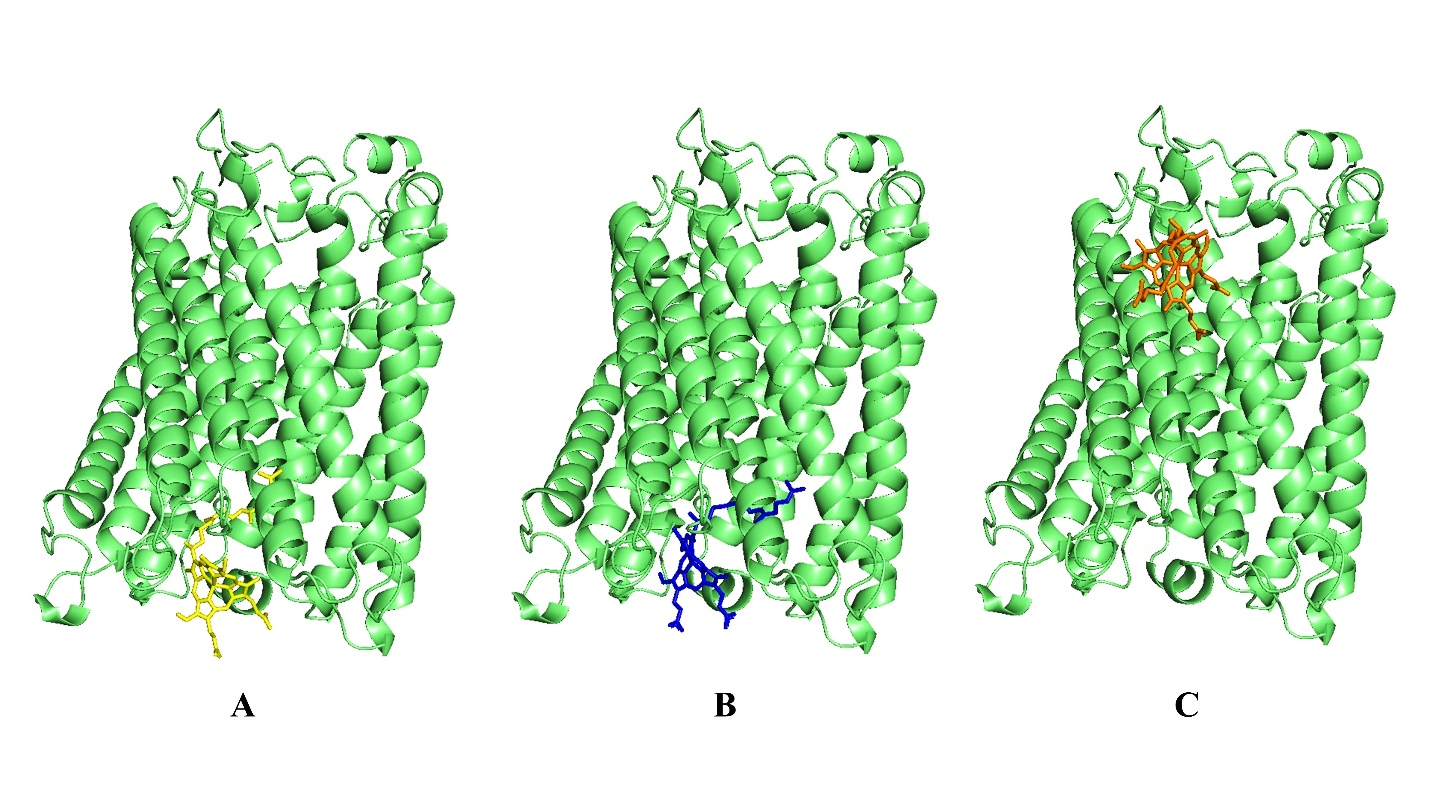
Figure 6. Molecular docking investigation of HP with Heme A. The site-specific docking was shown by the figure (A, B and C). Here, the ribbon that is displayed represents the HP and the strands depict the ligand (green color).
In our analysis, HP-HEA III has the lowest ligand RMSD value of 103.05 Å. The ligand and protein exhibited a substantial binding affinity. The analysis revealed a similarity between the HP and several interaction residues in the active site. The result aligns with the FTSite server's assessment of the active site. Results of the docking are illustrated in Figure 6 and Table 3.
Table 3. Investigation of molecular docking of the HP with the Heme A.
| Site ID | Interacting residues | Docking score | Confidence score | Ligand RMSD (Å) |
| HP-HEA I | HIS62, ALA63, MET66, ILE67, VAL71, TRP127, LEU247, VAL373, VAL374, PHE377, HIS378, LEU381, SER382, ARG438 | -108.99 | 0.3057 | 127.63 |
| HP-HEA II | SER33, ILE36, ARG37, LEU40, PHE55, VAL59, HIS62, TYR371, VAL374, ARG438, ARG439, ILE440 | -147.16 | 0.4858 | 127.43 |
| HP-HEA III | LEU17, MET20, LEU21, PHE24, PHE103, TRP104, PRO107 | -159.90 | 0.5493 | 103.05 |
In microbial and eukaryotic genomes, hypothetical proteins (HPs) are a significant although unexplored component. Bioinformatic characterization is crucial for identifying preliminary functions and directing future empirical studies because many of them lack experimental validation at the protein level, despite their presence being obvious at the sequence level [63]. Understanding the functions of HPs is essential to fully comprehending genomic and proteomic landscapes since these proteins often represent novel structures, undiscovered regulatory mechanisms, or uncharacterized enzymatic activity [21, 26]. Additionally, their characterization can shed light on metabolic pathways, mechanisms linked to disease, and possible targets for treatment [64].
In this study, we performed a comprehensive in silico evaluation of the hypothetical mitochondrial protein WN66_06778 from Saccharomyces cerevisiae. Physicochemical analysis revealed a stable, moderately acidic, hydrophobic protein with a calculated molecular weight of 58.75 kDa, a theoretical pI of 6.55, and a GRAVY score of 0.760. Multiple localization servers consistently predicted its mitochondrial residency, aligning with its hydrophobic nature and suggested transmembrane features. Secondary structure predictions indicated that the protein is predominantly alpha-helical, consistent with known structures of mitochondrial membrane proteins, particularly members of the electron transport chain [65, 66]. Functional annotation through InterProScan, Pfam, and NCBI-CDD strongly identified conserved motifs corresponding to cytochrome c oxidase subunit I (COX-I). This prediction was reinforced by BLAST-based homology analysis, which revealed up to 99% sequence identity with known COX-I proteins from closely related yeast species (Supplementary Table 2). COX-I is a core catalytic subunit of Complex IV, integral for proton pumping and electron transfer to molecular oxygen during oxidative phosphorylation. Its biogenesis is tightly coordinated through mitochondrial and nuclear gene expression, translational regulation, and cofactor incorporation [67-70].
Mitochondrial COX assembly requires an intricate interplay of nuclear-encoded factors, including Pet309, Mss51, and Shy1, which orchestrate COX-I translation, heme A insertion, and holoenzyme formation [ 71-76]. Disruption of these processes, often studied in petite mutants, impairs respiratory capacity and highlights the central role of COX-I as the structural scaffold for assembly of the full complex. Previous studies confirm that when COX subunits or cofactors are unavailable, COX-I synthesis is downregulated to prevent accumulation of assembly intermediates [73, 74]. The protein examined in this study demonstrated strong alignment with these conserved features, supporting its predicted classification as a COX-I-like protein.
Structural predictions generated using I-TASSER produced a high-confidence 3D model, which passed multiple validation criteria, including PROCHECK, QMEAN, ERRAT, and energy minimization via YASARA. The high TM-score (0.98 ± 0.05) indicates strong structural homology to experimentally resolved COX-I proteins. Active site mapping with FTSite identified approximately thirty putative functional residues, many of which aligned with known catalytic or ligand-binding positions within COX-I. Molecular docking with heme A, the primary cofactor for COX-I further substantiated this functional prediction. Among the docking complexes, HP-HEA III exhibited the strongest affinity (-159.90 kcal/mol) with superior confidence and lower RMSD values, indicating a stable and plausible interaction. Many interacting residues overlapped with the predicted active-site regions, reinforcing the protein’s likely catalytic capacity rather than a purely structural or accessory role [77-80].
The functional relevance of COX-I is underscored by its association with numerous mitochondrial pathologies. Defects in COX-I assembly or function can lead to impaired oxidative phosphorylation, elevated reactive oxygen species production, and severe clinical manifestations such as mitochondrial encephalomyopathies, Leigh syndrome, and neonatal-onset COX deficiency syndromes [81]. Several human orthologues of yeast COX assembly factors, including SCO1 and SCO2, are known contributors to such disorders, demonstrating evolutionary conservation and biomedical significance [82-84]. Thus, the accurate annotation of mitochondrial HPs with COX-I-like features may have broader implications for understanding mitochondrial dysfunction across species.
Overall, our findings offer strong computational evidence that the hypothetical protein WN66_06778 is a COX-I like mitochondrial protein with plausible catalytic activity and functional relevance within the electron transport chain. This study underscores the utility of integrative bioinformatics in clarifying the roles of uncharacterized proteins and sets the stage for targeted experimental validation. Beyond its biological importance in S. cerevisiae, this protein may hold translational relevance for mitochondrial research, industrial biotechnology, and the study of oxidative phosphorylation disorders.
This study provides a comprehensive computational characterization of the hypothetical mitochondrial protein WN66_06778 from Saccharomyces cerevisiae. Through integrated physicochemical analysis, structural modeling, domain annotation, evolutionary profiling, and molecular docking, we identified strong evidence that this uncharacterized protein represents a cytochrome c oxidase subunit I (COX-I) like protein. The predicted structural stability, predominant α-helical architecture, mitochondrial localization, and high sequence homology with known COX-I proteins collectively support this functional classification. High-confidence 3D modeling and validation further reinforced the reliability of the predicted structure, while binding site analysis and docking with heme A demonstrated robust ligand interactions consistent with a catalytic role in mitochondrial Complex IV. These findings not only clarify the probable biological role of WN66_06778 but also underscore its importance in oxidative phosphorylation and mitochondrial energy metabolism. Given the central role of COX-I in electron transport and the association of its dysfunction with diverse mitochondrial disorders, the detailed annotation of this protein creates a foundation for future experimental validation. Overall, this work highlights the power of in silico approaches for functional prediction of hypothetical proteins and provides valuable insights that may support future studies in yeast bioenergetics, mitochondrial biology, and industrial or biomedical applications.
The authors would like to thank the Faculty of Biotechnology and Genetic Engineering, Sylhet Agricultural University, Sylhet-3100, Bangladesh, and the Department of Biochemistry and Molecular Biology, Gono Bishwabidyalay (University), Dhaka-1344, Bangladesh, for the technical support of this research.
This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
The authors declare no conflicts of interest.
Not applicable.
The authors declare that artificial intelligence (AI) tools were used to assist in the preparation of this manuscript (approximately 10%) for improving language, grammar, and formatting. All content was reviewed and verified by the authors to ensure accuracy and compliance with ethical standards. The authors shall be solely responsible for any misconduct related to this work.
All supplementary materials referenced in this article are available in the online version.
 access
access